dynamics

Macromolecules exert their functions by dynamically changing their conformations. Taking advantage of the unique strength of the NMR techniques to provide quantitative dynamics information at atomic resolution, we have unveiled the function-related dynamics of biologically important systems. We have also developed an advanced structure-guided drug development (SGDD) strategy that actively utilizes the dynamics information for ligand optimization. The dynamic SGDD allows the development of ligands with preferable thermodynamics and promotes the utilization of new pharmaceutical modalities.
Dynamic ligand recognition by multidrug-binding proteins
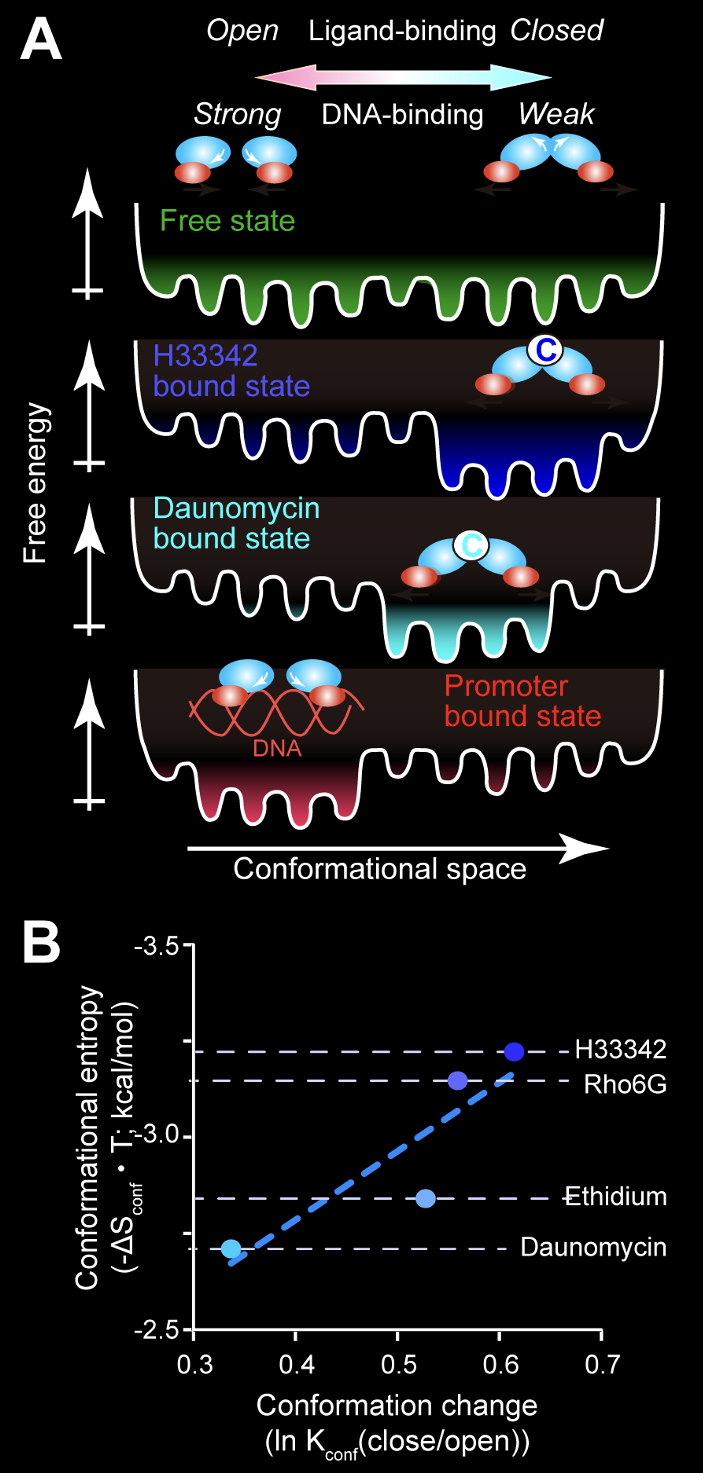

Multidrug resistance (MDR) systems are conserved in all three kingdoms of life, representing the most basic defense shared among organisms, and those of pathogenic bacteria and cancers are a major challenge for medication. However, the molecular mechanism by which multidrug-binding transcription factors (MDTF) recognize a variety of compounds with high affinity and regulate the transcription of MDR genes remains elusive.
By using NMR, we showed that an MDTF, LmrR, samples various conformations with different apertures in the ligand-binding site in the free state, while the conformations preferable to each ligand were selected upon the binding (Panel A, Sci. Rep., 2014; Sci. Rep., 2017). We also found that the regions allosteric to the ligand-binding site enhance the random fluctuation upon the binding to ligands, which entropically achieve a high-affinity interaction (Panel B). In addition, in the Staphylococcus aureus MDTF, QacR, the compound's size defines the induced activation levels through the fraction of the active conformation in the conformational equilibrium (Proc. Natl. Acad. Sci. USA, 2019). These results highlight the unique strength of NMR to unveil the dynamic molecular mechanism of proteins.
Dynamic activation of GPCR
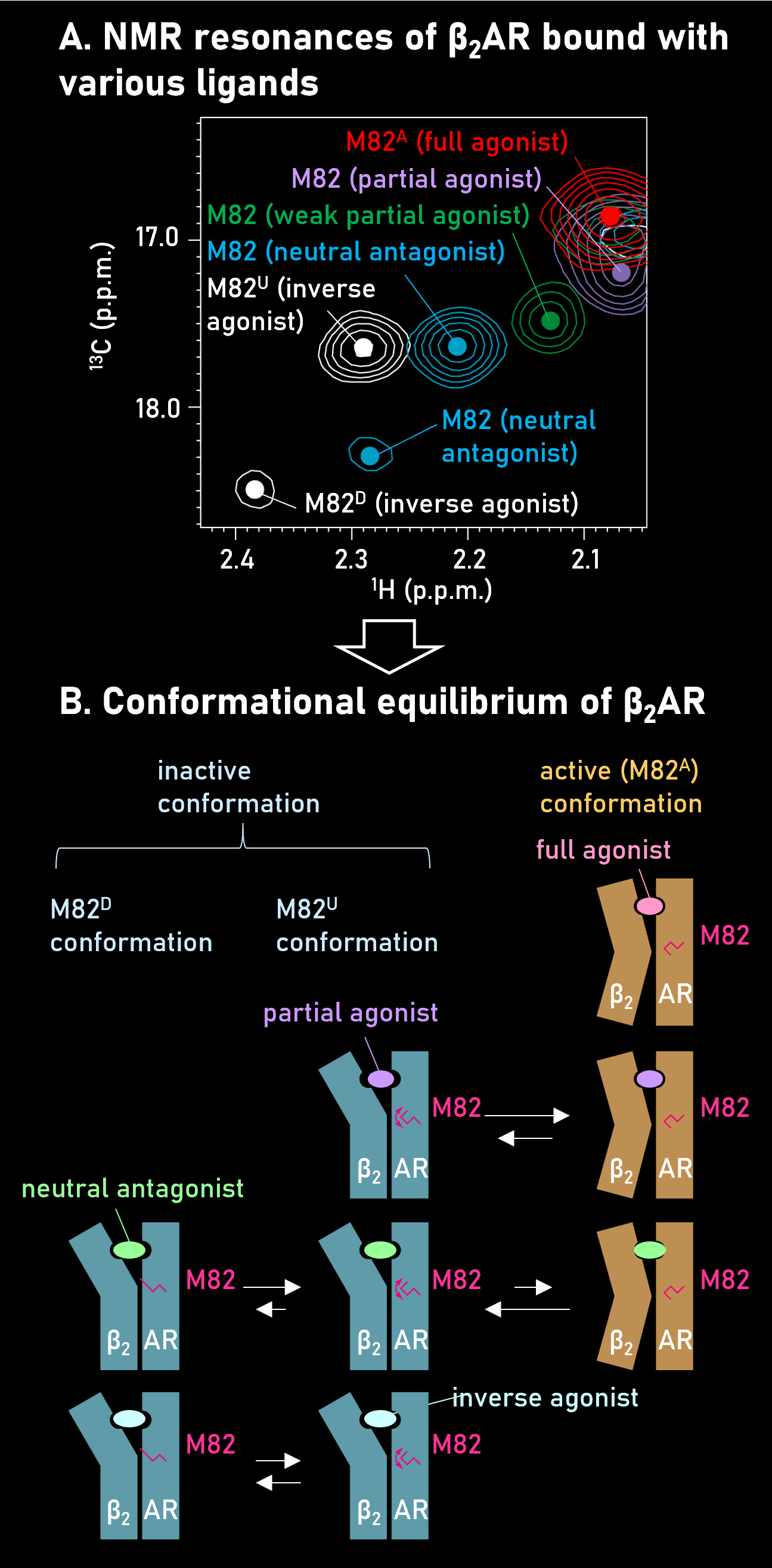
G protein-coupled receptors (GPCRs) are representative membrane proteins in eukaryotes, and induce various cellular responses upon the ligand binding. GPCRs are also a major drug target and one-third of the clinical drugs target GPCRs. The extent of intracellular activation evoked upon ligand binding is called efficacy, however, the molecular mechanism that defines the efficacy of each ligand has not been known.
By analyzing the β2-adrenergic receptor (β2AR) using NMR, we found that the signals from several transmembrane residues shift their positions in an efficacy-dependent manner(Panel A). The results indicate that the presence of an equilibrium between the active and inactive conformations in the transmembrane region of β2AR, and the population of active conformation defines the drug efficacy (panel B, Nat. Commun., 2012).
There are two types of GPCR signaling pathways, one mediated by G-proteins and the other by arrestin, and the balance between those two pathways is critical for the side effects of drugs. We showed that the dynamics of the transmembrane region define the balance of these two signaling pathways (Angew. Chem. Int. Ed., 2015). Such an investigation using NMR will lead to rational drug discovery that targets specific GPCRs.
NMR-based signal transduction model
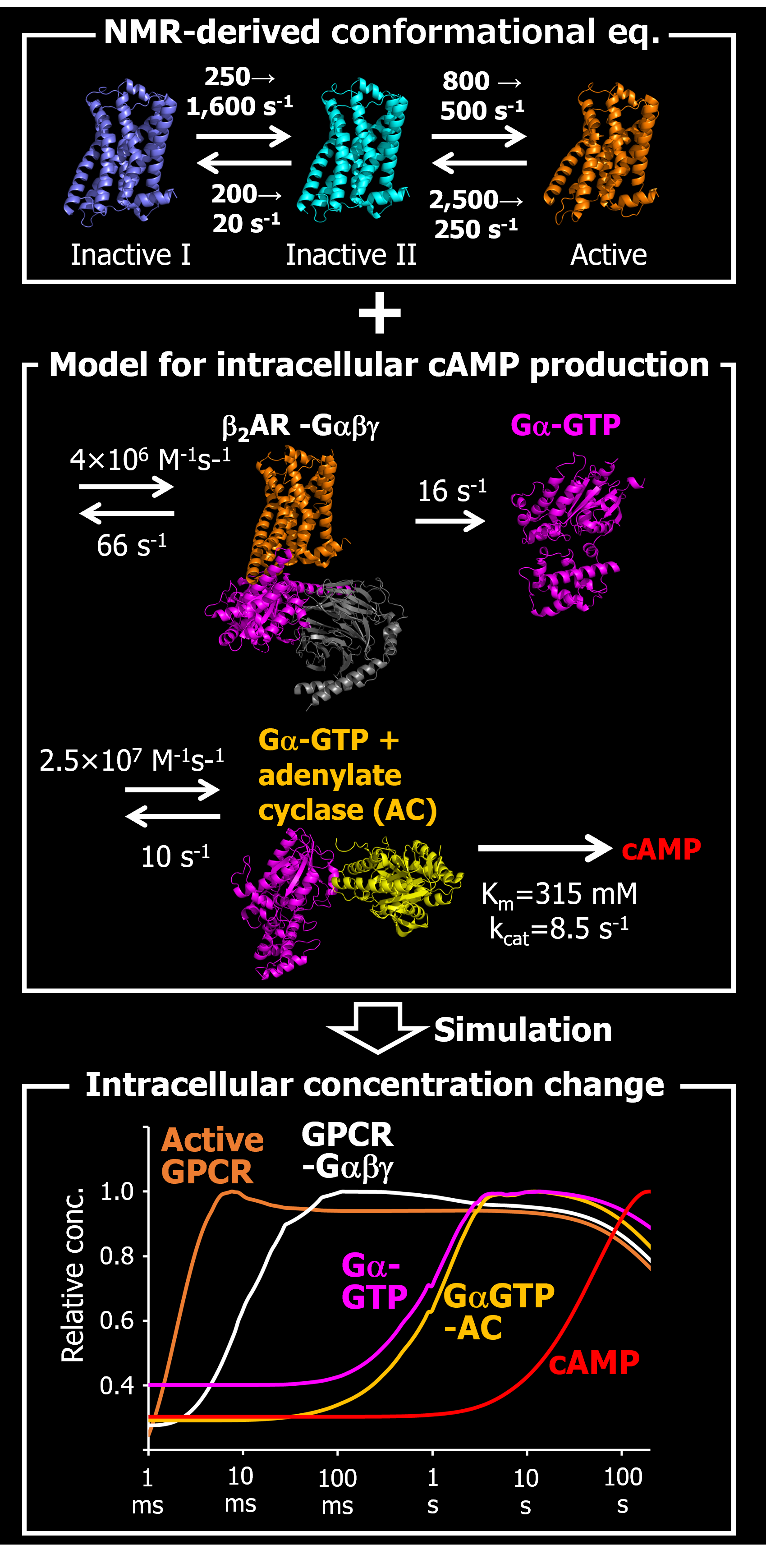
In living organism, proteins work in a concerted manner to exert physiological functions. Ligand binding alters the structure and dynamics of the receptor, which, in turn, changes its activity to exert a concerted function with other biomolecules. Therefore, it is necessary to construct a model that describes the entire picture of biological phenomena based on the protein dynamics information. In addition, in order to control a biological system, identification of the governing factor is of importance.
Toward this end, by integrating the NMR-derived function-related dynamics information with the mathematical model deduced from cell-based assays, the characteristic rate constants and temperature sensitivities of the GPCR signaling were successfully described by our strategy (Angew. Chem. Int. Ed., 2014; Sci. Rep., 2017). Although a complex mathematical model often requires enormously increasing computational resources, we avoided the problem by using the exchange Monte Carlo method, which exhaustively and efficiently explores a large number of parameters (Sci. Rep., 2017; Nat. Chem. Biol., 2020). We will further develop the strategy to elucidate the functional mechanisms that underlie various biological phenomena and to accelerate drug discovery.
Identification and utilization of cryptic sites
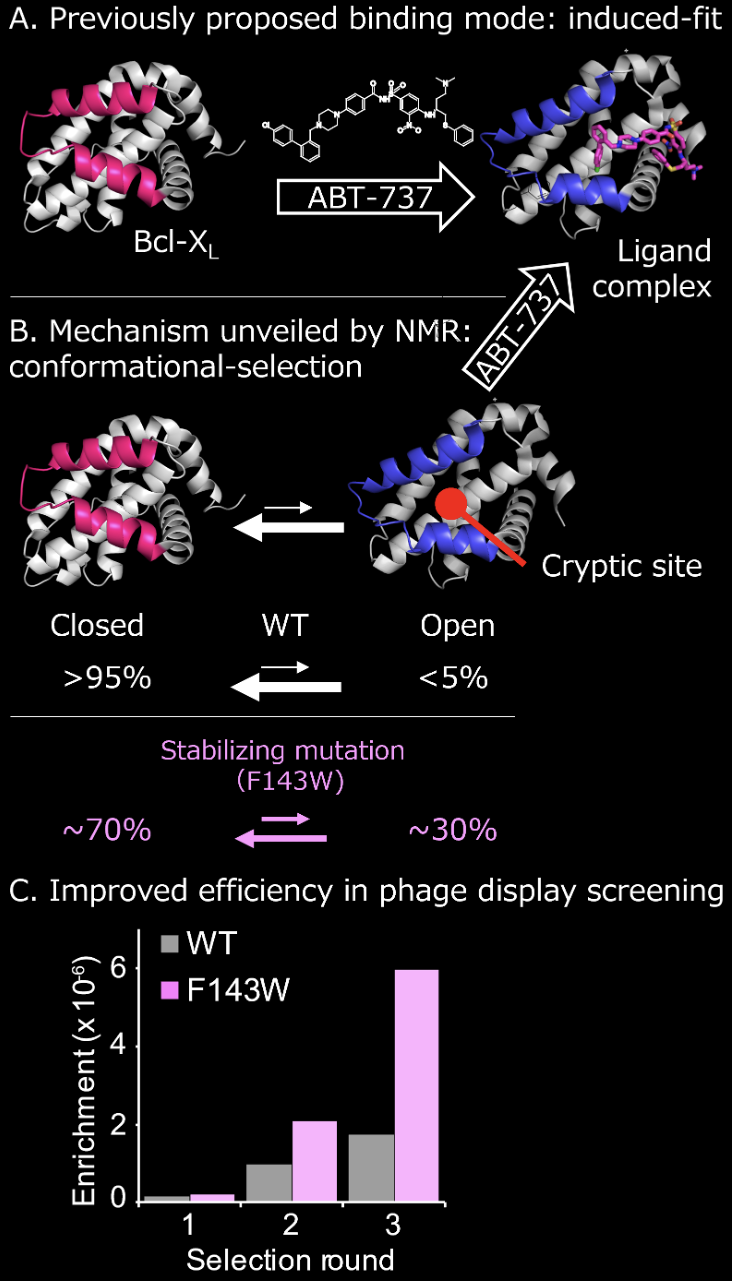
Protein-protein interactions (PPIs) contain many attractive drug targets. However, PPI-targeted drug discovery has been challenging since PPI sites are flat and wide and unsuitable for small molecules to bind. It is known that several successful PPI inhibitors bind to "cryptic sites" a dynamically formed ligand-interacting pocket that is not apparent in the absence of a ligand. Cryptic sites have been found only after discovering ligand and following structural analysis of the complexes (Panel A). However, if there is any strategy to discover and utilize cryptic sites in advance, we can accelerate the development of the PPI inhibitors and inhibitors of other difficult-to-target proteins.
By using NMR, we have found that a small fraction of cryptic sites already in the open conformation in Bcl-xL, an anti-cancer PPI inhibitor target, even in the absence of ligand (Panel B). We also obtained an allosteric mutant that stabilizes cryptic site open conformation and proved that the mutant could screen the hit compound more efficiently than the wild-type protein (Panel C). These results indicate that the druggability of proteins, including PPIs, can be improved by controlling the conformational equilibrium by using NMR. (Sci. Adv., 2019)