Publications
125.
Molecular basis promoting centriole triplet microtubule assembly.
2023
124.
Multimodal action of KRP203 on phosphoinositide kinases in vitro and in cells.
123.
The oxidative folding of nascent polypeptides provides electrons for reductive reactions in the ER.
122.
Nonthermal acceleration of protein hydration by sub-terahertz irradiation.
Nat Commun. 2023; 14(1):2825
121.
Amide-to-ester substitution as a stable alternative to N-methylation for increasing membrane permeability in cyclic peptides
120.
Functional molecular evolution of a GTP sensing kinase: PI5P4Kβ.
119.
Helix-forming aliphatic homo-δ-peptide foldamers based on the conformational restriction effects of cyclopropane.
118.
Absence of ULK1 decreases AMPK activity in the kidney, leading to chronic kidney disease progression.
2022
117.
Residue-based program of a β-peptoid twisted strand shape via a cyclopentane constraint.
Show abstractN-Substituted peptides, such as peptoids and β-peptoids, have been reported to have unique structures with diverse functions, like catalysis and manipulation of biomolecular functions. Recently, the preorganization of monomer shape by restricting bond rotations about all backbone dihedral angles has been demonstrated to be useful for de novo design of peptoid structures. Such design strategies are hitherto unexplored for β-peptoids; to date, no preorganized β-peptoid monomers have been reported. Here, we report the first design strategy for β-peptoids, in which all four backbone dihedral angles (ω, ϕ, θ, ψ) are rotationally restricted on a per-residue basis. The introduction of a cyclopentane constraint realized the preorganized monomer structure and led to a β-peptoid with a stable twisted strand shape.
116.
15N-Detected TROSY NMR experiments to study large disordered proteins in high-field magnets
Show abstractIntrinsically disordered regions (IDRs) of proteins are critical in the regulation of biological processes but difficult to study structurally. Nuclear magnetic resonance (NMR) is uniquely equipped to provide structural information on IDRs at atomic resolution; however, existing NMR methods often pose a challenge for large molecular weight IDRs. Resonance assignment of IDRs using 15ND-detection was previously demonstrated and shown to overcome some of these limitations. Here, we improve the methodology by overcoming the need for deuterated buffers and provide better sensitivity and resolution at higher magnetic fields and physiological salt concentrations using transverse relaxation optimized spectroscopy (TROSY). Finally, large disordered regions with low sequence complexity can be assigned efficiently using these new methods as demonstrated by achieving near complete assignment of the 398-residue N-terminal IDR of the transcription factor NFAT1 harboring 18% prolines.
115.
Repetitive DNA symmetry elements negatively regulate gene expression in embryonic stem cells
114.
The GTP responsiveness of PI5P4Kβ evolved from a compromised trade-off between activity and specificity.
Show abstract & figure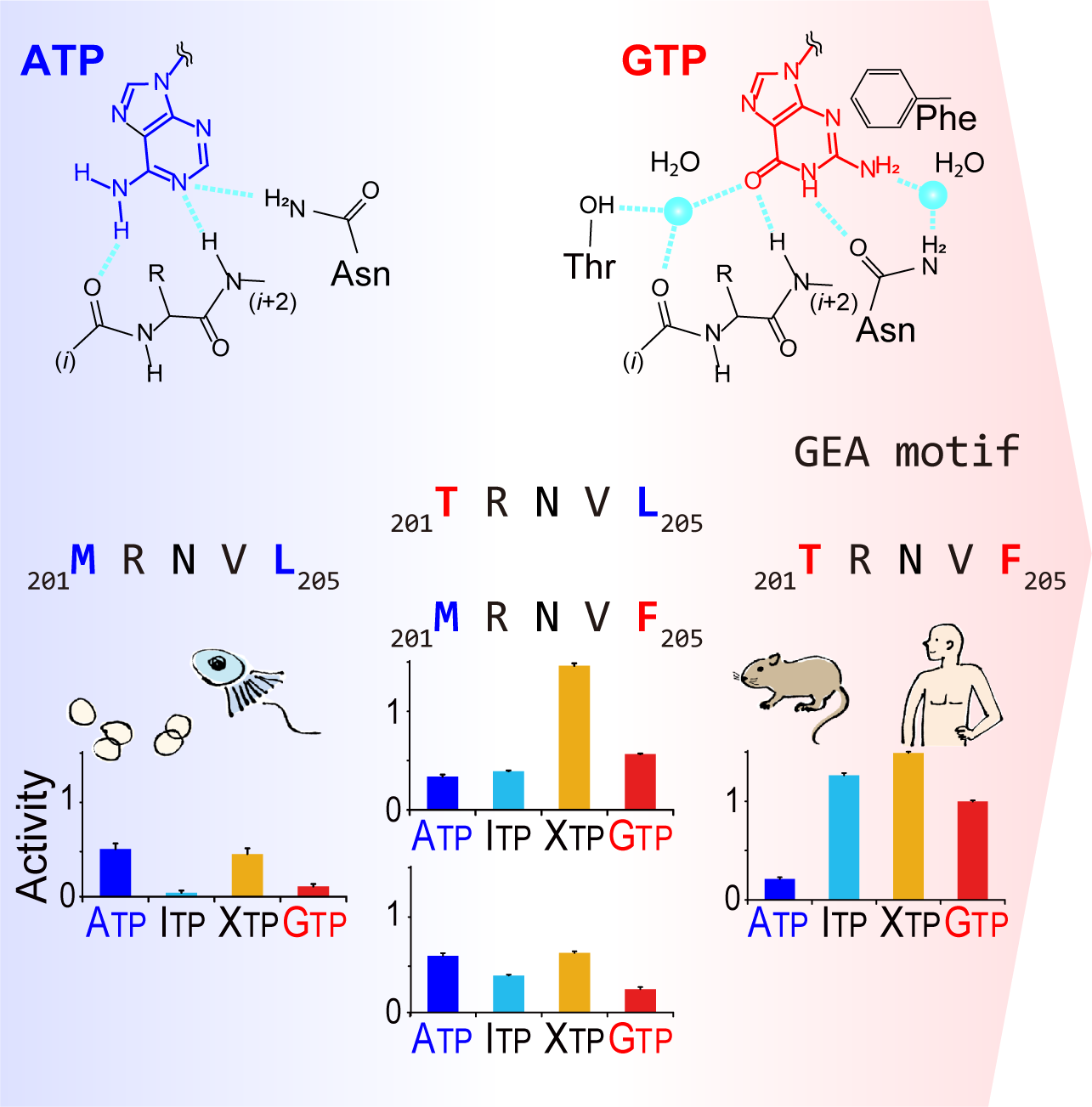

Unlike most kinases, phosphatidylinositol 5-phosphate 4-kinase β (PI5P4Kβ) utilizes GTP as a physiological phosphate donor and regulates cell growth under stress (i.e., GTP-dependent stress resilience). However, the genesis and evolution of its GTP responsiveness remain unknown. Here, we reveal that PI5P4Kβ has acquired GTP preference by generating a short dual-nucleotide-recognizing motif called the guanine efficient association (GEA) motif. Comparison of nucleobase recognition with 660 kinases and 128 G proteins has uncovered that most kinases and PI5P4Kβ use their main-chain atoms for adenine recognition, while the side-chain atoms are required for guanine recognition. Mutational analysis of the GEA motif revealed that the acquisition of GTP reactivity is accompanied by an extended activity toward inosine triphosphate (ITP) and xanthosine triphosphate (XTP). Along with the evolutionary analysis data that point to strong negative selection of the GEA motif, these results suggest that the GTP responsiveness of PI5P4Kβ has evolved from a compromised trade-off between activity and specificity, underpinning the development of the GTP-dependent stress resilience.
113.
Role of the Orphan Transporter SLC35E1 in the Nuclear Egress of Herpes Simplex Virus 1
112.
Oligo(N-methylalanine) as a Peptide-Based Molecular Scaffold with a Minimal Structure and High Density of Functionalizable Sites
Show abstractFunctionalizable synthetic molecules with nanometer sizes and defined shapes in water are useful as molecular scaffolds to mimic the functions of biomacromolecules and develop chemical tools for manipulating biomacromolecules. Herein, we propose oligo(N-methylalanine) (oligo-NMA) as a peptide-based molecular scaffold with a minimal structure and a high density of functionalizable sites. Oligo-NMA forms a defined shape in water without hydrogen-bonding networks or ring constraints, which enables the molecule to act as a scaffold with minimal atomic composition. Furthermore, functional groups can be readily introduced on the nitrogens and α-carbons of oligo-NMA. Computational and NMR spectroscopic analysis suggested that the backbone structure of oligo-NMA is not largely affected by functionalization. Moreover, the usefulness of oligo-NMA was demonstrated by the design of protein ligands. The ease of synthesis, minimal structure, and high functionalization flexibility makes oligo-NMA a useful scaffold for chemical and biological applications.
2021
111.
Biphasic activation of β-arrestin 1 upon interaction with a GPCR revealed by methyl-TROSY NMR.
Nat Commun 2021; 12(1):7158
Show abstractβ-arrestins (βarrs) play multifaceted roles in the function of G protein-coupled receptors (GPCRs). βarrs typically interact with phosphorylated C-terminal tail (C tail) and transmembrane core (TM core) of GPCRs. However, the effects of the C tail- and TM core-mediated interactions on the conformational activation of βarrs have remained elusive. Here, we show the conformational changes for βarr activation upon the C tail- and TM core-mediated interactions with a prototypical GPCR by nuclear magnetic resonance (NMR) spectroscopy. Our NMR analyses demonstrated that while the C tail-mediated interaction alone induces partial activation, in which βarr exists in equilibrium between basal and activated conformations, the TM core- and the C tail-mediated interactions together completely shift the equilibrium toward the activated conformation. The conformation-selective antibody, Fab30, promotes partially activated βarr into the activated-like conformation. This plasticity of βarr conformation in complex with GPCRs engaged in different binding modes may explain the multifunctionality of βarrs.
110.
Function-Related Dynamics in Multi-Spanning Helical Membrane Proteins Revealed by Solution NMR.
Show abstractA primary biological function of multi-spanning membrane proteins is to transfer information and/or materials through a membrane by changing their conformations. Therefore, particular dynamics of the membrane proteins are tightly associated with their function. The semi-atomic resolution dynamics information revealed by NMR is able to discriminate function-related dynamics from random fluctuations. This review will discuss several studies in which quantitative dynamics information by solution NMR has contributed to revealing the structural basis of the function of multi-spanning membrane proteins, such as ion channels, GPCRs, and transporters.
109.
3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents.
108.
Divergent Mechanisms Activating RAS and Small GTPases Through Post-translational Modification.
Show abstract & figure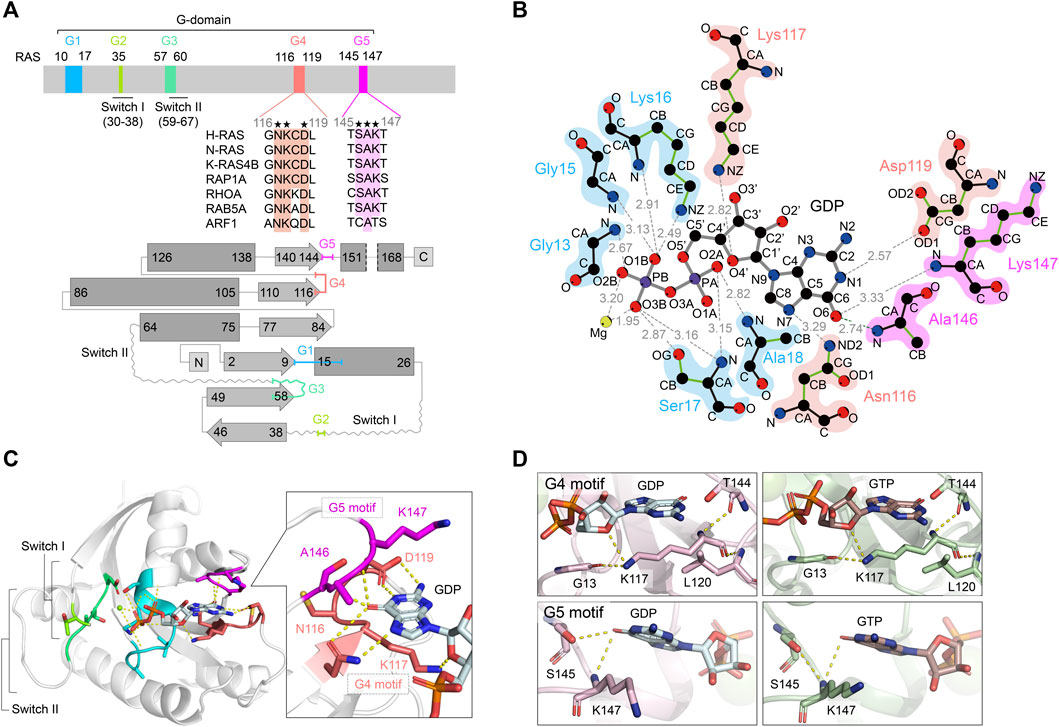
RAS is a founding member of the RAS superfamily of GTPases. These small 21 kDa proteins function as molecular switches to initialize signaling cascades involved in various cellular processes, including gene expression, cell growth, and differentiation. RAS is activated by GTP loading and deactivated upon GTP hydrolysis to GDP. Guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) accelerate GTP loading and hydrolysis, respectively. These accessory proteins play a fundamental role in regulating activities of RAS superfamily small GTPase via a conserved guanine binding (G)-domain, which consists of five G motifs. The Switch regions lie within or proximal to the G2 and G3 motifs, and undergo dynamic conformational changes between the GDP-bound "OFF" state and GTP-bound "ON" state. They play an important role in the recognition of regulatory factors (GEFs and GAPs) and effectors. The G4 and G5 motifs are the focus of the present work and lie outside Switch regions. These motifs are responsible for the recognition of the guanine moiety in GTP and GDP, and contain residues that undergo post-translational modifications that underlie new mechanisms of RAS regulation. Post-translational modification within the G4 and G5 motifs activates RAS by populating the GTP-bound "ON" state, either through enhancement of intrinsic guanine nucleotide exchange or impairing GAP-mediated down-regulation. Here, we provide a comprehensive review of post-translational modifications in the RAS G4 and G5 motifs, and describe the role of these modifications in RAS activation as well as potential applications for cancer therapy.
107.
Nonthermal excitation effects mediated by sub-terahertz radiation on hydrogen exchange in ubiquitin.
106.
Flexibility and Cell Permeability of Cyclic Ras-Inhibitor Peptides Revealed by the Coupled Nosé-Hoover Equation.
105.
Conformational Plasticity of Cyclic Ras-Inhibitor Peptides Defines Cell Permeabilization Activity.
Angew Chem Int Ed Engl 2021; 60(12):6567-6572
Show abstract & figure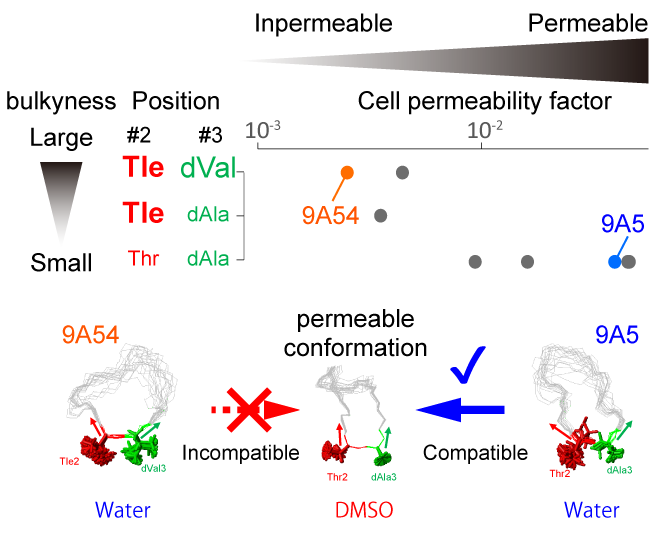
Cyclorasins 9A5 and 9A54 are 11-mer cyclic peptides that inhibit the Ras-Raf protein interaction. The peptides share a cell-penetrating peptide (CPP)-like motif; however, only cyclorasin 9A5 can permeabilize cells to exhibit strong cell-based activity. To unveil the structural origin underlying their distinct cellular permeabilization activities, we compared the three-dimensional structures of cyclorasins 9A5 and 9A54 in water and in the less polar solvent dimethyl sulfoxide (DMSO) by solution NMR. We found that cyclorasin 9A5 changes its extended conformation in water to a compact amphipathic structure with converged aromatic residues surrounded by Arg residues in DMSO, which might contribute to its cell permeabilization activity. However, cyclorasin 9A54 cannot adopt this amphipathic structure, due to the steric hindrance between two neighboring bulky amino-acid sidechains, Tle-2 and dVal-3. We also found that the bulkiness of the sidechains at positions 2 and 3 negatively affects the cell permeabilization activities, indicating that the conformational plasticity that allows the peptides to form the amphipathic structure is important for their cell permeabilization activities.
2020
104.
Role of NMR in High Ordered Structure Characterization of Monoclonal Antibodies.
Show abstractObtaining high ordered structure (HOS) information is of importance to guarantee the efficacy and safety of monoclonal antibodies (mAbs) in clinical application. Assessment of HOS should ideally be performed in a non-invasive manner under their formulated storage conditions, as any perturbation can introduce unexpected detritions. However, most of the currently available techniques only indirectly report HOS of mAbs and/or require a certain condition to conduct the analyses. Besides, the flexible multidomain architecture of mAbs has hampered atomic-resolution structural analyses using X-ray crystallography and cryo-electron microscopy. In contrast, the ability of nuclear magnetic resonance (NMR) spectroscopy to structurally analyze biomolecules in various conditions in a non-invasive and quantitative manner is suitable to meet the needs. However, the application of NMR to mAbs is not straightforward due to the high molecular weight of the system. In this review, we will discuss how NMR techniques have been applied to HOS analysis of mAbs, along with the recent advances of the novel 15N direct detection NMR strategy that allows for obtaining the structural fingerprint of mAbs at lower temperatures under multiple formulation conditions. The potential application of these NMR strategies will benefit next-generation mAbs, such as antibody-drug conjugates and bispecific antibodies.
103.
The role of NMR in leveraging dynamics and entropy in drug design.
102.
Targeting the cryptic sites: NMR-based strategy to improve protein druggability by controlling the conformational equilibrium.
Sci Adv 2020; 6(40):eabd0480
Show abstract & figure
Cryptic ligand binding sites, which are not evident in the unligated structures, are beneficial in tackling with difficult but attractive drug targets, such as protein-protein interactions (PPIs). However, cryptic sites have thus far not been rationally pursued in the early stages of drug development. Here, we demonstrated by nuclear magnetic resonance that the cryptic site in Bcl-xL exists in a conformational equilibrium between the open and closed conformations under the unligated condition. While the fraction of the open conformation in the unligated wild-type Bcl-xL is estimated to be low, F143W mutation that is distal from the ligand binding site can substantially elevate the population. The F143W mutant showed a higher hit rate in a phage-display peptide screening, and the hit peptide bound to the cryptic site of the wild-type Bcl-xL. Therefore, by controlling the conformational equilibrium in the cryptic site, the opportunity to identify a PPI inhibitor could be improved.
101.
Control of Transcription Initiation by Biased Thermal Fluctuations on Repetitive Genomic Sequences.
Show abstractIn the process of transcription initiation by RNA polymerase, promoter DNA sequences affect multiple reaction pathways determining the productivity of transcription. However, the question of how the molecular mechanism of transcription initiation depends on the sequence properties of promoter DNA remains poorly understood. Here, combining the statistical mechanical approach with high-throughput sequencing results, we characterize abortive transcription and pausing during transcription initiation by Escherichia coli RNA polymerase at a genome-wide level. Our results suggest that initially transcribed sequences, when enriched with thymine bases, contain the signal for inducing abortive transcription, whereas certain repetitive sequence elements embedded in promoter regions constitute the signal for inducing pausing. Both signals decrease the productivity of transcription initiation. Based on solution NMR and in vitro transcription measurements, we suggest that repetitive sequence elements within the promoter DNA modulate the nonlocal base pair stability of its double-stranded form. This stability profoundly influences the reaction coordinates of the productive initiation via pausing.
100.
Mutation-induced change in chignolin stability from π-turn to α-turn
RSC Adv 2020; 10(38):22797-22808
Show abstractChignolin, which consists of 10 amino acids, adopts two stable states in simulations at room temperature at 1 atm: the native and misfolded states. The sequence of chignolin is optimized to form a stable π-turn and thus the native state has a π-turn from Asp3 to Thr8. On the other hand, the misfolded state adopts an α-turn from Asp3 to Gly7. We previously investigated the differences in the stability mechanism of the two states using computational techniques. Our previous detailed energy analysis implied that the native state was stabilized by hydrogen bonding between the side chain atoms of Thr6 and Thr8, and Thr8 was not involved in stabilization of the misfolded state. Thus, we predicted that mutation of Thr8 to a neutral amino acid could stabilize the misfolded structure over the native structure. In the present work, we performed 4 μs molecular dynamics simulations for 19 mutants of the 8th residue. Among them, the T8I, T8F, T8P, T8N, and T8Y mutants, in which the 8th residue was changed to a neutral residue, formed only the misfolded structure at room temperature. Even at high temperature, for the T8P mutant, the native structure was not observed, as the T8P mutant cannot form the native structure because of steric hindrance caused by the distinctive cyclic structure of proline. Interestingly, the T8P mutant at high temperature has trans and cis conformations in the Gly7–Pro8 sequence, with the trans conformation corresponding to the misfolded state. NMR analysis of the T8P mutant supported our results.
99.
Structural Fingerprints of an Intact Monoclonal Antibody Acquired under Formulated Storage Conditions via 15N Direct Detection Nuclear Magnetic Resonance.
98.
Transcription Factor Binding in Embryonic Stem Cells Is Constrained by DNA Sequence Repeat Symmetry.
97.
Structural equilibrium underlying ligand-dependent activation of β2-adrenoreceptor.
Nat Chem Biol 2020; 16(4):430-439
Show abstractG-protein-coupled receptors (GPCRs) are seven-transmembrane proteins mediating cellular signals in response to extracellular stimuli. Although three-dimensional structures showcase snapshots that can be sampled in the process and nuclear magnetic resonance detects conformational equilibria, the mechanism by which agonist-activated GPCRs interact with various effectors remains elusive. Here, we used paramagnetic nuclear magnetic resonance for leucine amide resonances to visualize the structure of β2-adrenoreceptor in the full agonist-bound state, without thermostabilizing mutations abolishing its activity. The structure exhibited a unique orientation of the intracellular half of the transmembrane helix 6, forming a cluster of G-protein-interacting residues. Furthermore, analyses of efficacy-dependent chemical shifts of the residues near the pivotal PIF microswitch identified an equilibrium among three conformations, including one responsible for the varied signal level in each ligand-bound state. Together, these results provide a structural basis for the dynamic activation of GPCRs and shed light on GPCR-mediated signal transduction.
96.
Activation of adenosine A2A receptor by lipids from docosahexaenoic acid revealed by NMR.
Sci Adv 2020; 6(12):eaay8544
Show abstractThe lipid composition of the plasma membrane is a key parameter in controlling signal transduction through G protein-coupled receptors (GPCRs). Adenosine A2A receptor (A2AAR) is located in the lipid bilayers of cells, containing acyl chains derived from docosahexaenoic acid (DHA). For the NMR studies, we prepared A2AAR in lipid bilayers of nanodiscs, containing DHA chains and other acyl chains. The DHA chains in nanodiscs enhanced the activation of G proteins by A2AAR. Our NMR studies revealed that the DHA chains redistribute the multiple conformations of A2AAR toward those preferable for G protein binding. In these conformations, the rotational angle of transmembrane helix 6 is similar to that in the A2AAR-G protein complex, suggesting that the population shift of the equilibrium causes the enhanced activation of G protein by A2AAR. These findings provide insights into the control of neurotransmissions by A2AAR and the effects of lipids on various GPCR functions.
95.
Spotlight on the Ballet of Proteins: The Structural Dynamic Properties of Proteins Illuminated by Solution NMR.
Show abstractSolution NMR spectroscopy is a unique and powerful technique that has the ability to directly connect the structural dynamics of proteins in physiological conditions to their activity and function. Here, we summarize recent studies in which solution NMR contributed to the discovery of relationships between key dynamic properties of proteins and functional mechanisms in important biological systems. The capacity of NMR to quantify the dynamics of proteins over a range of time scales and to detect lowly populated protein conformations plays a critical role in its power to unveil functional protein dynamics. This analysis of dynamics is not only important for the understanding of biological function, but also in the design of specific ligands for pharmacologically important proteins. Thus, the dynamic view of structure provided by NMR is of importance in both basic and applied biology.
94.
Targeting FROUNT with disulfiram suppresses macrophage accumulation and its tumor-promoting properties.
Show abstractTumor-associated macrophages affect tumor progression and resistance to immune checkpoint therapy. Here, we identify the chemokine signal regulator FROUNT as a target to control tumor-associated macrophages. The low level FROUNT expression in patients with cancer correlates with better clinical outcomes. Frount-deficiency markedly reduces tumor progression and decreases macrophage tumor-promoting activity. FROUNT is highly expressed in macrophages, and its myeloid-specific deletion impairs tumor growth. Further, the anti-alcoholism drug disulfiram (DSF) acts as a potent inhibitor of FROUNT. DSF interferes with FROUNT-chemokine receptor interactions via direct binding to a specific site of the chemokine receptor-binding domain of FROUNT, leading to inhibition of macrophage responses. DSF monotherapy reduces tumor progression and decreases macrophage tumor-promoting activity, as seen in the case of Frount-deficiency. Moreover, co-treatment with DSF and an immune checkpoint antibody synergistically inhibits tumor growth. Thus, inhibition of FROUNT by DSF represents a promising strategy for macrophage-targeted cancer therapy.
93.
Prediction of Passive Membrane Permeability by Semi-Empirical Method Considering Viscous and Inertial Resistances and Different Rates of Conformational Change and Diffusion.
Show abstractMembrane permeability is an important property of drugs in adsorption. Many prediction methods work well for small molecules, but the prediction of middle-molecule permeability is still difficult. In the present study, we modified a classical permeability model based on Fick's law to study passive membrane permeability. The model consisted of the distribution of solute from water to membrane and the diffusion of solute in each solvent. The diffusion coefficient is the inverse of the resistance, and we examined the inertial resistance in addition to the viscous resistance, the latter of which has been widely used in permeability prediction. Also, we examined three models changing the balance between the diffusion of solute in membrane and the conformational change of solute. The inertial resistance improved the prediction results in addition to the viscous resistance. The models worked well not only for small molecules but also for middle molecules, whose structures have more conformational freedom.
2019
92.
A BRET-based assay reveals collagen-Hsp47 interaction dynamics in the endoplasmic reticulum and small-molecule inhibition of this interaction.
Show abstractMolecular chaperones perform pivotal roles in proteostasis by engaging in protein-protein interactions (PPIs). The collagen-specific molecular chaperone Hsp47 (heat shock protein 47) interacts with procollagen in the endoplasmic reticulum (ER) and plays crucial roles in collagen synthesis. PPIs between Hsp47 and collagen could offer a therapeutic target for fibrosis, which is characterized by abnormal collagen accumulation in the extracellular matrix of fibrotic organs. Herein, we established a bioluminescence resonance energy transfer (BRET) system for assessing Hsp47-collagen interaction dynamics within the ER. After optimization and validation of the method, we could demonstrate inhibition of the interaction between Hsp47 and collagen by a small molecule (Col003) in the ER. Using the BRET system, we also found that Hsp47 interacts not only with the Gly-Pro-Arg motif but also weakly with Gly-Pro-Hyp motifs of triple-helical collagen in cells. Moreover, we found that the serpin loop of Hsp47 (SerpinH1) contributes to its binding to collagen. We propose that the method developed here can provide valuable information on PPIs between Hsp47 and collagen and on the effects of PPI inhibitors important for the management of fibrotic disorders.
91.
Conformational equilibrium defines the variable induction of the multidrug-binding transcriptional repressor QacR.
Show abstractQacR, a multidrug-binding transcriptional repressor in pathogenic bacteria Staphylococcus aureus, modulates the transcriptional level of the multidrug transporter gene, qacA, in response to engaging a set of diverse ligands. However, the structural basis that defines the variable induction level remains unknown. Here, we reveal that the conformational equilibrium between the repressive and inducive conformations in QacR defines the induction level of the transporter gene. In addition, the unligated QacR is already partly populated in the inducive conformation, allowing the basal expression of the transporter. We also showed that, in the known constitutively active QacR mutants, the equilibrium is shifted more toward the inducive conformation, even in the unligated state. These results highlight the unexpected structural mechanism, connecting the promiscuous multidrug binding to the variable transcriptional regulation of QacR, which provide clues to dysfunctioning of the multidrug resistance systems.
90.
Emerging solution NMR methods to illuminate the structural and dynamic properties of proteins.
Show abstractThe first recognition of protein breathing was more than 50 years ago. Today, we are able to detect the multitude of interaction modes, structural polymorphisms, and binding-induced changes in protein structure that direct function. Solution-state NMR spectroscopy has proved to be a powerful technique, not only to obtain high-resolution structures of proteins, but also to provide unique insights into the functional dynamics of proteins. Here, we summarize recent technical landmarks in solution NMR that have enabled characterization of key biological macromolecular systems. These methods have been fundamental to atomic resolution structure determination and quantitative analysis of dynamics over a wide range of time scales by NMR. The ability of NMR to detect lowly populated protein conformations and transiently formed complexes plays a critical role in its ability to elucidate functionally important structural features of proteins and their dynamics.
89.
Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story.
Show abstractThe purine nucleotides ATP and GTP are essential precursors to DNA and RNA synthesis and fundamental for energy metabolism. Although de novo purine nucleotide biosynthesis is increased in highly proliferating cells, such as malignant tumors, it is not clear if this is merely a secondary manifestation of increased cell proliferation. Suggestive of a direct causative effect includes evidence that, in some cancer types, the rate-limiting enzyme in de novo GTP biosynthesis, inosine monophosphate dehydrogenase (IMPDH), is upregulated and that the IMPDH inhibitor, mycophenolic acid (MPA), possesses anti-tumor activity. However, historically, enthusiasm for employing IMPDH inhibitors in cancer treatment has been mitigated by their adverse effects at high treatment doses and variable response. Recent advances in our understanding of the mechanistic role of IMPDH in tumorigenesis and cancer progression, as well as the development of IMPDH inhibitors with selective actions on GTP synthesis, have prompted a reappraisal of targeting this enzyme for anti-cancer treatment. In this review, we summarize the history of IMPDH inhibitors, the development of new inhibitors as anti-cancer drugs, and future directions and strategies to overcome existing challenges.
88.
Structure determination using solution NMR: Is it worth the effort?
Show abstractIt has been almost 40 years since solution NMR joined X-ray crystallography as a technique for determining high-resolution structures of proteins. Since then NMR derived structure has contributed in fundamental ways to our understanding of the function of biomolecules. With the already existing mature field of X-ray crystallography and the emergence of cryo-EM as techniques to tackle high-resolution structures of large protein complexes, the role of NMR in structure determination has been questioned. However, NMR has the unique ability to recapitulate the dynamic motion of proteins in their structures, while size limitations of the biomolecular systems that can be routinely studied still present challenges. The field has continually developed methodology and instrumentation since its introduction, pushing its frontiers and redefining its limits. Here we present a brief overview of NMR-based structure determination over the past 40 years. We outline the current state of the field and look ahead to the challenges that still need to be addressed to realize the future potential of NMR as a structural technique.
87.
Function-related conformational dynamics of G protein-coupled receptors revealed by NMR.
Show abstractG protein-coupled receptors (GPCRs) function as receptors for various neurotransmitters, hormones, cytokines, and metabolites. GPCR ligands impart differing degrees of signaling in the G protein and arrestin pathways, in phenomena called biased signaling, and each ligand for a given GPCR has a characteristic level of ability to activate or deactivate its target, which is referred to as its efficacy. The ligand efficacies and biased signaling of GPCRs remarkably affect the therapeutic properties of the ligands. However, these features of GPCRs can only be partially understood from the crystallography data, although numerous GPCR structures have been solved. NMR analyses have revealed that GPCRs have multiple interconverting substates, exchanging on various timescales, and that the exchange rates are related to the ligand efficacies and biased signaling. In addition, NMR analyses of GPCRs in the lipid bilayer environment of rHDLs revealed that the exchange rates are modulated by the lipid bilayer environment, highlighting the importance of the function-related dynamics in the lipid bilayer. In this review, we will describe several solution NMR studies that have clarified the conformational dynamics related to the ligand efficacy and biased signaling of GPCRs.
86.
Aromatic 19F-13C TROSY: a background-free approach to probe biomolecular structure, function, and dynamics.
Show abstract & figure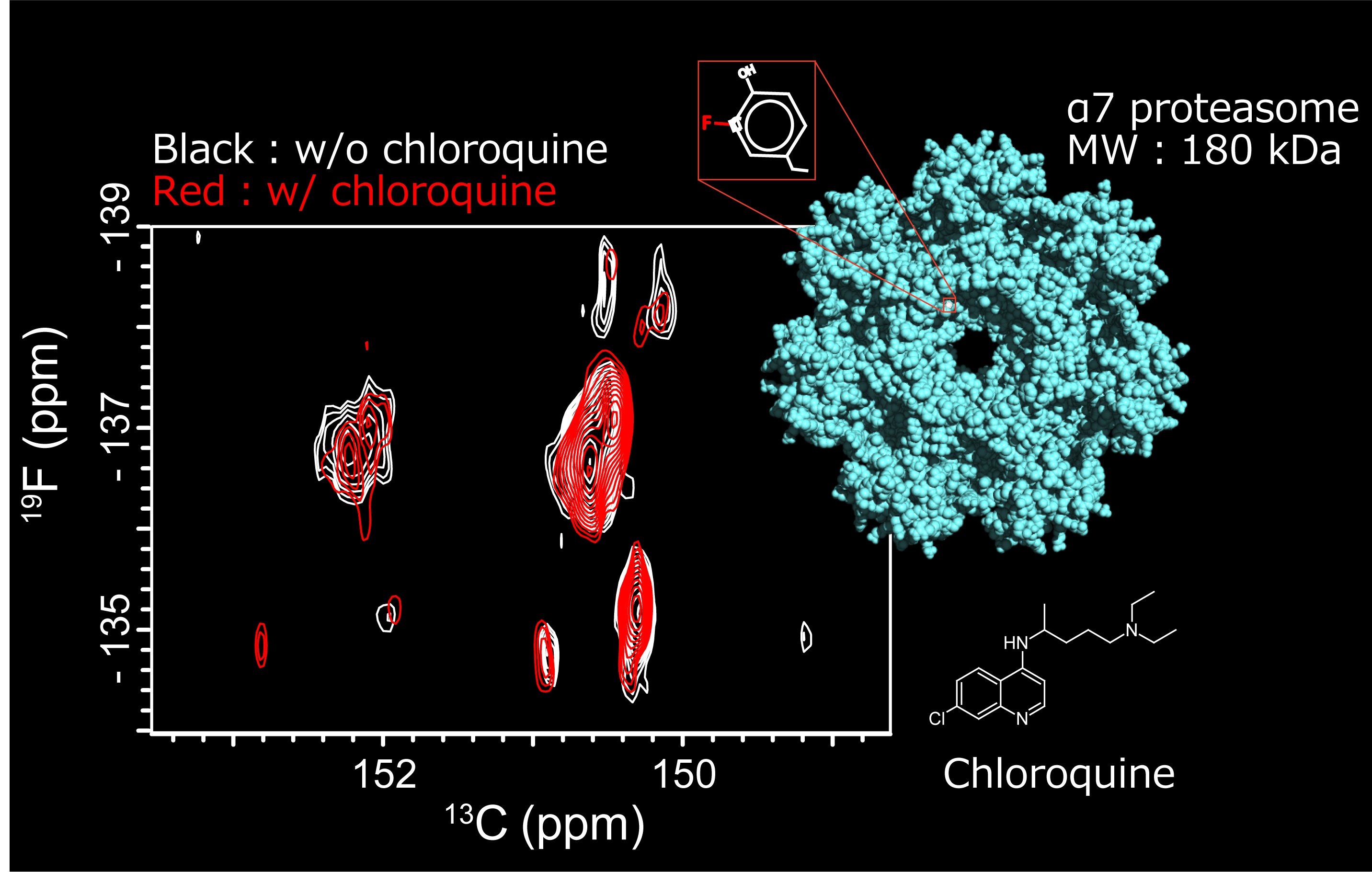
Atomic-level information about the structure and dynamics of biomolecules is critical for an understanding of their function. Nuclear magnetic resonance (NMR) spectroscopy provides unique insights into the dynamic nature of biomolecules and their interactions, capturing transient conformers and their features. However, relaxation-induced line broadening and signal overlap make it challenging to apply NMR spectroscopy to large biological systems. Here we took advantage of the high sensitivity and broad chemical shift range of 19F nuclei and leveraged the remarkable relaxation properties of the aromatic 19F-13C spin pair to disperse 19F resonances in a two-dimensional transverse relaxation-optimized spectroscopy spectrum. We demonstrate the application of 19F-13C transverse relaxation-optimized spectroscopy to investigate proteins and nucleic acids. This experiment expands the scope of 19F NMR in the study of the structure, dynamics, and function of large and complex biological systems and provides a powerful background-free NMR probe.
85.
Screening for inhibitor of episomal DNA identified dicumarol as a hepatitis B virus inhibitor.
Show abstractCurrently, there is no available therapy to eradicate hepatitis B virus (HBV) in chronically infected individuals. This is due to the difficulty in eliminating viral covalently closed circular (ccc) DNA, which is central to the gene expression and replication of HBV. We developed an assay system for nuclear circular DNA using an integration-deficient lentiviral vector. This vector produced non-integrated circular DNA in nuclei of infected cells. We engineered this vector to encode firefly luciferase to monitor the lentiviral episome DNA. We screened 3,840 chemicals by this assay for luciferase-reducing activity and identified dicumarol, which is known to have anticoagulation activity. We confirmed that dicumarol reduced lentiviral episome DNA. Furthermore, dicumarol inhibited HBV replication in cell culture using NTCP-expressing HepG2 and primary human hepatocytes. Dicumarol reduced intracellular HBV RNA, DNA, supernatant HBV antigens and DNA. We also found that dicumarol reduced the cccDNA level in HBV infected cells, but did not affect HBV adsorption/entry. This is a novel assay system for screening inhibitors targeting nuclear cccDNA and is useful for finding new antiviral substances for HBV.
84.
GPCR drug discovery: integrating solution NMR data with crystal and cryo-EM structures.
2018
83.
Application of C-Terminal 7-Azabicyclo[2.2.1]heptane to Stabilize β-Strand-like Extended Conformation of a Neighboring α-Amino Acid.
82.
Competing protein-protein interactions regulate binding of Hsp27 to its client protein tau.
Show abstractSmall heat shock proteins (sHSPs) are a class of oligomeric molecular chaperones that limit protein aggregation. However, it is often not clear where sHSPs bind on their client proteins or how these protein-protein interactions (PPIs) are regulated. Here, we map the PPIs between human Hsp27 and the microtubule-associated protein tau (MAPT/tau). We find that Hsp27 selectively recognizes two aggregation-prone regions of tau, using the conserved β4-β8 cleft of its alpha-crystallin domain. The β4-β8 region is also the site of Hsp27-Hsp27 interactions, suggesting that competitive PPIs may be an important regulatory paradigm. Indeed, we find that each of the individual PPIs are relatively weak and that competition for shared sites seems to control both client binding and Hsp27 oligomerization. These findings highlight the importance of multiple, competitive PPIs in the function of Hsp27 and suggest that the β4-β8 groove acts as a tunable sensor for clients.
81.
Phosphoinositide binding by the PH domain in ceramide transfer protein (CERT) is inhibited by hyperphosphorylation of an adjacent serine-repeat motif.
Show abstractSphingolipids such as ceramide are important constituents of cell membranes. The ceramide transfer protein (CERT) moves ceramide from the endoplasmic reticulum to the Golgi apparatus in a nonvesicular manner. Hyperphosphorylation of the serine-repeat motif (SRM) adjacent to the pleckstrin homology (PH) domain of CERT down-regulates the inter-organelle ceramide transport function of CERT. However, the mechanistic details of this down-regulation remain elusive. Using solution NMR and binding assays, we herein show that a hyperphosphorylation-mimetic CERT variant in which 10 serine/threonine residues of SRM had been replaced with glutamate residues (the 10E variant) displays an intramolecular interaction between SRM and positively charged regions of the PH domain, which are involved in the binding of this domain to phosphatidylinositol 4-monophosphate (PI4P). Of note, the binding of the PH domain to PI4P-embedded membranes was attenuated by the SRM 10E substitutions in cell-free assays. Moreover, the 10E substitutions reduced the Golgi-targeting activity of the PH-SRM construct in living cells. These results indicate that hyperphosphorylated SRM directly interacts with the surface of the PH domain in an intramolecular manner, thereby decreasing the PI4P-binding activity of the PH domain. In light of these findings, we propose that the hyperphosphorylation of SRM may trigger the dissociation of CERT from the Golgi apparatus, resulting in a functionally less active conformation of CERT.
80.
Deuteration and selective labeling of alanine methyl groups of β2-adrenergic receptor expressed in a baculovirus-insect cell expression system.
79.
15N detection harnesses the slow relaxation property of nitrogen: Delivering enhanced resolution for intrinsically disordered proteins.
Show abstractStudies over the past decade have highlighted the functional significance of intrinsically disordered proteins (IDPs). Due to conformational heterogeneity and inherent dynamics, structural studies of IDPs have relied mostly on NMR spectroscopy, despite IDPs having characteristics that make them challenging to study using traditional 1H-detected biomolecular NMR techniques. Here, we develop a suite of 3D 15N-detected experiments that take advantage of the slower transverse relaxation property of 15N nuclei, the associated narrower linewidth, and the greater chemical shift dispersion compared with those of 1H and 13C resonances. The six 3D experiments described here start with aliphatic 1H magnetization to take advantage of its higher initial polarization, and are broadly applicable for backbone assignment of proteins that are disordered, dynamic, or have unfavorable amide proton exchange rates. Using these experiments, backbone resonance assignments were completed for the unstructured regulatory domain (residues 131-294) of the human transcription factor nuclear factor of activated T cells (NFATC2), which includes 28 proline residues located in functionally important serine-proline (SP) repeats. The complete assignment of the NFATC2 regulatory domain enabled us to study phosphorylation of NFAT by kinase PKA and phosphorylation-dependent binding of chaperone protein 14-3-3 to NFAT, providing mechanistic insight on how 14-3-3 regulates NFAT nuclear translocation.
78.
Mixed pyruvate labeling enables backbone resonance assignment of large proteins using a single experiment.
Show abstractBackbone resonance assignment is a critical first step in the investigation of proteins by NMR. This is traditionally achieved with a standard set of experiments, most of which are not optimal for large proteins. Of these, HNCA is the most sensitive experiment that provides sequential correlations. However, this experiment suffers from chemical shift degeneracy problems during the assignment procedure. We present a strategy that increases the effective resolution of HNCA and enables near-complete resonance assignment using this single HNCA experiment. We utilize a combination of 2-13C and 3-13C pyruvate as the carbon source for isotope labeling, which suppresses the one bond (1Jαβ) coupling providing enhanced resolution for the Cα resonance and amino acid-specific peak shapes that arise from the residual coupling. Using this approach, we can obtain near-complete (>85%) backbone resonance assignment of a 42 kDa protein using a single HNCA experiment.
77.
Phosphorylation-induced conformation of β2-adrenoceptor related to arrestin recruitment revealed by NMR.
Nat Commun 2018; 9(1):194
Show abstractThe C-terminal region of G-protein-coupled receptors (GPCRs), stimulated by agonist binding, is phosphorylated by GPCR kinases, and the phosphorylated GPCRs bind to arrestin, leading to the cellular responses. To understand the mechanism underlying the formation of the phosphorylated GPCR-arrestin complex, we performed NMR analyses of the phosphorylated β2-adrenoceptor (β2AR) and the phosphorylated β2AR-β-arrestin 1 complex, in the lipid bilayers of nanodisc. Here we show that the phosphorylated C-terminal region adheres to either the intracellular side of the transmembrane region or lipids, and that the phosphorylation of the C-terminal region allosterically alters the conformation around M2155.54 and M2796.41, located on transemembrane helices 5 and 6, respectively. In addition, we found that the conformation induced by the phosphorylation is similar to that corresponding to the β-arrestin-bound state. The phosphorylation-induced structures revealed in this study propose a conserved structural motif of GPCRs that enables β-arrestin to recognize dozens of GPCRs.
2017
76.
A small-molecule compound inhibits a collagen-specific molecular chaperone and could represent a potential remedy for fibrosis.
Show abstractFibrosis can disrupt tissue structure and integrity and impair organ function. Fibrosis is characterized by abnormal collagen accumulation in the extracellular matrix. Pharmacological inhibition of collagen secretion therefore represents a promising strategy for the management of fibrotic disorders, such as liver and lung fibrosis. Hsp47 is an endoplasmic reticulum (ER)-resident collagen-specific molecular chaperone essential for correct folding of procollagen in the ER. Genetic deletion of Hsp47 or inhibition of its interaction with procollagen interferes with procollagen triple helix production, which vastly reduces procollagen secretion from fibroblasts. Thus, Hsp47 could be a potential and promising target for the management of fibrosis. In this study, we screened small-molecule compounds that inhibit the interaction of Hsp47 with collagen from chemical libraries using surface plasmon resonance (BIAcore), and we found a molecule AK778 and its cleavage product Col003 competitively inhibited the interaction and caused the inhibition of collagen secretion by destabilizing the collagen triple helix. Structural information obtained with NMR analysis revealed that Col003 competitively binds to the collagen-binding site on Hsp47. We propose that these structural insights could provide a basis for designing more effective therapeutic drugs for managing fibrosis.
75.
15N-Detection with TROSY Selection Enables the Study of Large Nondeuterated Macromolecular Systems
eMagRes 2017; 6(3):369-380
74.
Forbidden Coherence Transfer of 19F Nuclei to Quantitatively Measure the Dynamics of a CF₃-Containing Ligand in Receptor-Bound States.
Show abstractThe dynamic property of a ligand in the receptor-bound state is an important metric to characterize the interactions in the ligand-receptor interface, and the development of an experimental strategy to quantify the amplitude of motions in the bound state is of importance to introduce the dynamic aspect into structure-guided drug development (SGDD). Fluorine modifications are frequently introduced at the hit-to-lead optimization stage to enhance the binding potency and other characteristics of a ligand. However, the effects of fluorine modifications are generally difficult to predict, owing to the pleiotropic nature of the interactions. In this study, we report an NMR-based approach to experimentally evaluate the local dynamics of trifluoromethyl (CF₃)-containing ligands in the receptor-bound states. For this purpose, the forbidden coherence transfer (FCT) analysis, which has been used to study the dynamics of methyl moieties in proteins, was extended to the 19F nuclei of CF₃-containing ligands. By applying this CF₃-FCT analysis to a model interaction system consisting of a ligand, AST-487, and a receptor, p38α, we successfully quantified the amplitude of the CF₃ dynamics in the p38α-bound state. The strategy would bring the CF₃-containing ligands within the scope of dynamic SGDD to improve the affinity and specificity for the drug-target receptors.
73.
The crystal structure of a new O-demethylase from Sphingobium sp. strain SYK-6.
Show abstractIn the cell, tetrahydrofolate (H4 folate) derivatives with a C1 unit are utilized in various ways, such as for the synthesis of amino acids and nucleic acids. While H4 folate derivatives with the C1 unit are typically produced in the glycine cleavage system, Sphingobium sp. strain SYK-6, which can utilize lignin-derived aromatic compounds as a sole source of carbon and energy, lacks this pathway, probably due to its unique nutrient requirements. In this bacterium, H4 folate-dependent O-demethylases in catabolic pathways for lignin-derived aromatic compounds seem to be involved in the C1 metabolism. LigM is one of the O-demethylases and catalyzes a C1-unit transfer from vanillate (VNL) to H4 folate. As the primary structure of LigM shows a similarity to T-protein in the glycine cleavage system, we hypothesized that LigM has evolved from T-protein, acquiring its unique biochemical and biological functions. To prove this hypothesis, structure-based understanding of its catalytic reaction is essential. Here, we determined the crystal structure of LigM in apo form and in complex with substrates and H4 folate. These crystal structures showed that the overall structure of LigM is similar to T-protein, but LigM has a few distinct characteristics, particularly in the active site. Structure-based mutational analysis revealed that His60 and Tyr247, which are not conserved in T-protein, are essential to the catalytic activity of LigM and their interactions with the oxygen atom in the methoxy group of VNL seem to facilitate a methyl moiety (C1-unit) transfer to H4 folate. Taken together, our structural data suggest that LigM has evolved divergently from T-protein.
72.
Dynamic equilibrium on DNA defines transcriptional regulation of a multidrug binding transcriptional repressor, LmrR.
Show abstractLmrR is a multidrug binding transcriptional repressor that controls the expression of a major multidrug transporter, LmrCD, in Lactococcus lactis. Promiscuous compound ligations reduce the affinity of LmrR for the lmrCD operator by several fold to release the transcriptional repression; however, the affinity reduction is orders of magnitude smaller than that of typical transcriptional repressors. Here, we found that the transcriptional regulation of LmrR is achieved through an equilibrium between the operator-bound and non-specific DNA-adsorption states in vivo. The effective dissociation constant of LmrR for the lmrCD operator under the equilibrium is close to the endogenous concentration of LmrR, which allows a substantial reduction of LmrR occupancy upon compound ligations. Therefore, LmrR represents a dynamic type of transcriptional regulation of prokaryotic multidrug resistance systems, where the small affinity reduction induced by compounds is coupled to the functional relocalization of the repressor on the genomic DNA via nonspecific DNA adsorption.
71.
Quantitative Structure-activity Relationship (QSAR) Models for Docking Score Correction.
Show abstractIn order to improve docking score correction, we developed several structure-based quantitative structure activity relationship (QSAR) models by protein-drug docking simulations and applied these models to public affinity data. The prediction models used descriptor-based regression, and the compound descriptor was a set of docking scores against multiple (∼600) proteins including nontargets. The binding free energy that corresponded to the docking score was approximated by a weighted average of docking scores for multiple proteins, and we tried linear, weighted linear and polynomial regression models considering the compound similarities. In addition, we tried a combination of these regression models for individual data sets such as IC50 , Ki , and %inhibition values. The cross-validation results showed that the weighted linear model was more accurate than the simple linear regression model. Thus, the QSAR approaches based on the affinity data of public databases should improve docking scores.
2016
70.
Perspective: revisiting the field dependence of TROSY sensitivity.
69.
Improvement of Ligand Affinity and Thermodynamic Properties by NMR-Based Evaluation of Local Dynamics and Surface Complementarity in the Receptor-Bound State.
Show abstractThe thermodynamic properties of a ligand in the bound state affect its binding specificity. Strict binding specificity can be achieved by introducing multiple spatially defined interactions, such as hydrogen bonds and van der Waals interactions, into the ligand-receptor interface. These introduced interactions are characterized by restricted local dynamics and improved surface complementarity in the bound state. In this study, we experimentally evaluated the local dynamics and the surface complementarity of weak-affinity ligands in the receptor-bound state by forbidden coherence transfer analysis in free-bound exchange systems (Ex-FCT), using the interaction between a ligand, a myocyte-enhancer factor 2A (MEF2A) docking peptide, and a receptor, p38α, as a model system. The Ex-FCT analyses successfully provided information for the rational design of a ligand with higher affinity and preferable thermodynamic properties for p38α.
68.
Structural reverse genetics study of the PI5P4Kβ-nucleotide complexes reveals the presence of the GTP bioenergetic system in mammalian cells.
Show abstract & figure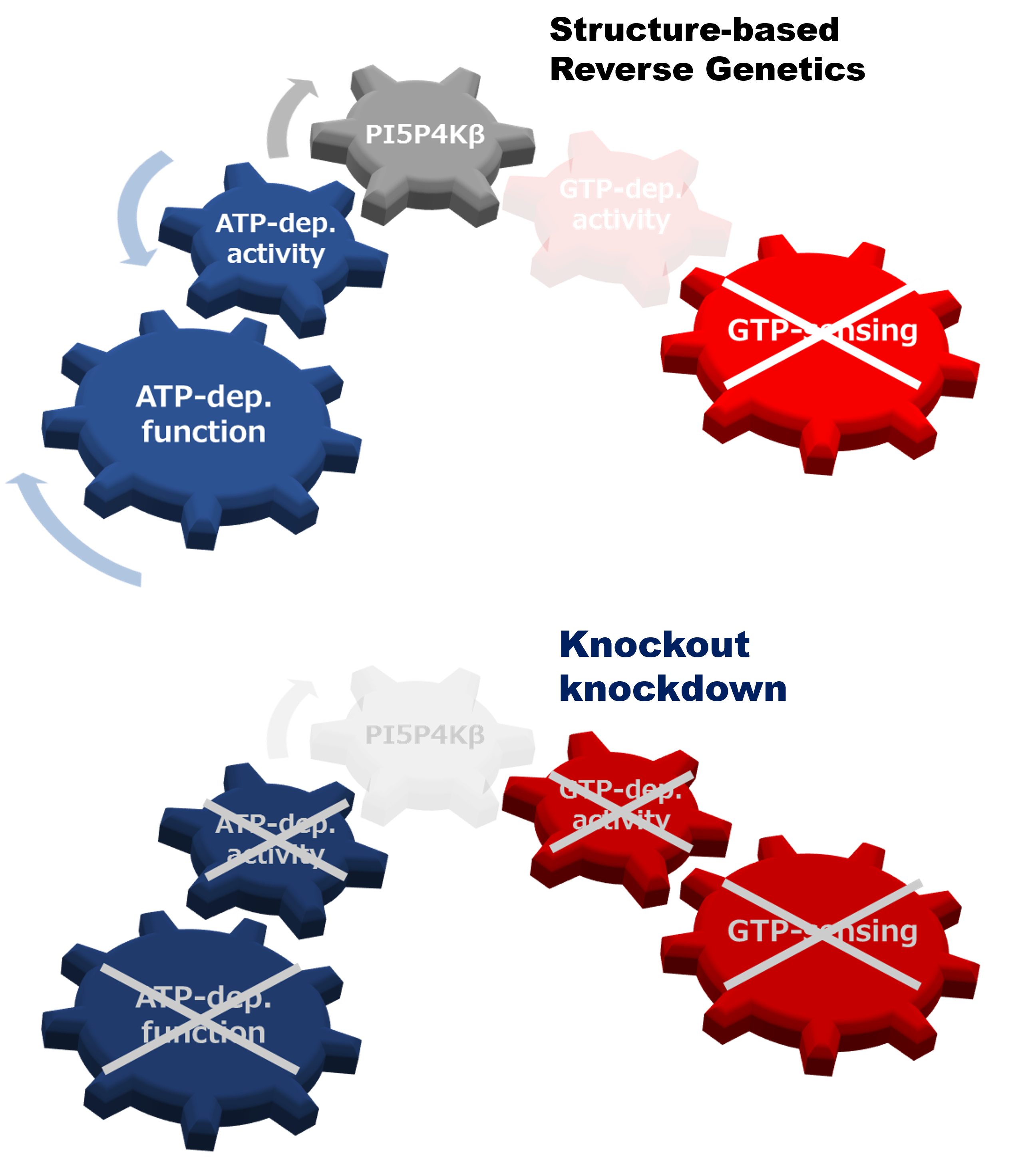
Reverse genetic analysis can connect a gene and its protein counterpart to a biological function(s) by knockout or knockdown of the specific gene. However, when a protein has multiple biochemical activities, the conventional genetics strategy is incapable of distinguishing which biochemical activity of the protein is critical for the particular biological function(s). Here, we propose a structural reverse genetics strategy to overcome this problem. In a structural reverse genetics study, multiple biochemical activities of a protein are segregated by mapping those activities to a structural element(s) in the atomic resolution tertiary structure. Based on the structural mapping, a mutant lacking one biochemical activity of interest can be produced with the other activities kept intact. Expression of the mutant by knockin or ectopic expression in the knockout strain along with the following analysis can connect the single biochemical activity of interest to a biological function. Using the structural reverse genetics strategy, we have dissected the newly identified GTP-dependent activity of a lipid kinase PI5P4Kβ from its ATP-dependent activity. The GTP-insensitive mutant has demonstrated the existence of the GTP bioenergetic sensor system in mammalian cells and its critical role in tumorigenesis. As structural reverse genetics can identify in vivo significance of individual biochemical activity, it is a powerful approach to reveal hidden biological functions, which could be a novel pharmacological target for therapeutic intervention. Given the recent expansion of choices in structural biological methods and advances in genome editing technologies, the time is ripe for structural reverse genetics strategies.
67.
Utilization of paramagnetic relaxation enhancements for structural analysis of actin-binding proteins in complex with actin.
Show abstractActin cytoskeleton dynamics are controlled by various actin binding proteins (ABPs) that modulate the polymerization of the monomeric G-actin and the depolymerization of filamentous F-actin. Although revealing the structures of the actin/ABP complexes is crucial to understand how the ABPs regulate actin dynamics, the X-ray crystallography and cryoEM methods are inadequate to apply for the ABPs that interact with G- or F-actin with lower affinity or multiple binding modes. In this study, we aimed to establish the alternative method to build a structural model of G-actin/ABP complexes, utilizing the paramagnetic relaxation enhancement (PRE) experiments. Thymosin β4 (Tβ4) was used as a test case for validation, since its structure in complex with G-actin was reported recently. Recombinantly expressed G-actin, containing a cysteine mutation, was conjugated with a nitroxyl spin label at the specific site. Based on the intensity ratio of the 1H-15N HSQC spectra of Tβ4 in the complex with G-actin in the paramagnetic and diamagnetic states, the distances between the amide groups of Tβ4 and the spin label of G-actin were estimated. Using the PRE-derived distance constraints, we were able to compute a well-converged docking structure of the G-actin/Tβ4 complex that shows great accordance with the reference structure.
66.
Conductance of P2X4 purinergic receptor is determined by conformational equilibrium in the transmembrane region.
Show abstractLigand-gated ion channels are partially activated by their ligands, resulting in currents lower than the currents evoked by the physiological full agonists. In the case of P2X purinergic receptors, a cation-selective pore in the transmembrane region expands upon ATP binding to the extracellular ATP-binding site, and the currents evoked by α,β-methylene ATP are lower than the currents evoked by ATP. However, the mechanism underlying the partial activation of the P2X receptors is unknown although the crystal structures of zebrafish P2X4 receptor in the apo and ATP-bound states are available. Here, we observed the NMR signals from M339 and M351, which were introduced in the transmembrane region, and the endogenous alanine and methionine residues of the zebrafish P2X4 purinergic receptor in the apo, ATP-bound, and α,β-methylene ATP-bound states. Our NMR analyses revealed that, in the α,β-methylene ATP-bound state, M339, M351, and the residues that connect the ATP-binding site and the transmembrane region, M325 and A330, exist in conformational equilibrium between closed and open conformations, with slower exchange rates than the chemical shift difference (<100 s(-1)), suggesting that the small population of the open conformation causes the partial activation in this state. Our NMR analyses also revealed that the transmembrane region adopts the open conformation in the state bound to the inhibitor trinitrophenyl-ATP, and thus the antagonism is due to the closure of ion pathways, except for the pore in the transmembrane region: i.e., the lateral cation access in the extracellular region.
65.
Nitrogen-detected TROSY yields comparable sensitivity to proton-detected TROSY for non-deuterated, large proteins under physiological salt conditions.
Show abstractDirect detection of the TROSY component of proton-attached (15)N nuclei ((15)N-detected TROSY) yields high quality spectra with high field magnets, by taking advantage of the slow (15)N transverse relaxation. The slow transverse relaxation and narrow line width of the (15)N-detected TROSY resonances are expected to compensate for the inherently low (15)N sensitivity. However, the sensitivity of (15)N-detected TROSY in a previous report was one-order of magnitude lower than in the conventional (1)H-detected version. This could be due to the fact that the previous experiments were performed at low salt (0-50 mM), which is advantageous for (1)H-detected experiments. Here, we show that the sensitivity gap between (15)N and (1)H becomes marginal for a non-deuterated, large protein (τ c = 35 ns) at a physiological salt concentration (200 mM). This effect is due to the high salt tolerance of the (15)N-detected TROSY. Together with the previously reported benefits of the (15)N-detected TROSY, our results provide further support for the significance of this experiment for structural studies of macromolecules when using high field magnets near and above 1 GHz.
64.
Use of Multiple Cryoprotectants to Improve Diffraction Quality from Protein Crystals
Cryst Growth Des 2016; 16(3):1565-1571
63.
The Lipid Kinase PI5P4Kβ Is an Intracellular GTP Sensor for Metabolism and Tumorigenesis.
Show abstract & figure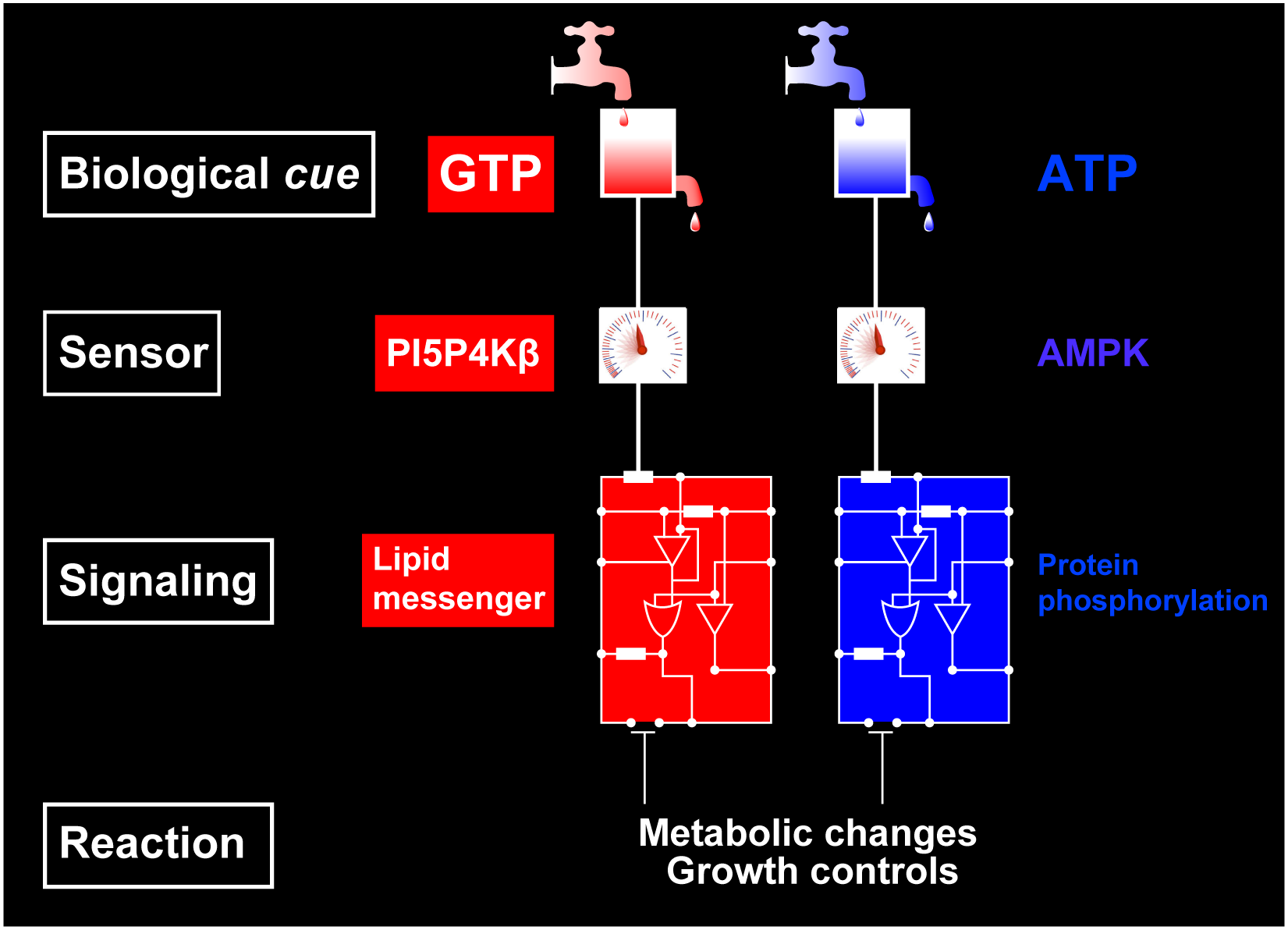
While cellular GTP concentration dramatically changes in response to an organism's cellular status, whether it serves as a metabolic cue for biological signaling remains elusive due to the lack of molecular identification of GTP sensors. Here we report that PI5P4Kβ, a phosphoinositide kinase that regulates PI(5)P levels, detects GTP concentration and converts them into lipid second messenger signaling. Biochemical analyses show that PI5P4Kβ preferentially utilizes GTP, rather than ATP, for PI(5)P phosphorylation, and its activity reflects changes in direct proportion to the physiological GTP concentration. Structural and biological analyses reveal that the GTP-sensing activity of PI5P4Kβ is critical for metabolic adaptation and tumorigenesis. These results demonstrate that PI5P4Kβ is the missing GTP sensor and that GTP concentration functions as a metabolic cue via PI5P4Kβ. The critical role of the GTP-sensing activity of PI5P4Kβ in cancer signifies this lipid kinase as a cancer therapeutic target.
2015
62.
Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor.
Show abstractG-protein-coupled receptor (GPCR) ligands impart differing degrees of signaling in the G-protein and arrestin pathways, in phenomena called "biased signaling". However, the mechanism underlying the biased signaling of GPCRs is still unclear, although crystal structures of GPCRs bound to the G protein or arrestin are available. In this study, we observed the NMR signals from methionine residues of the μ-opioid receptor (μOR) in the balanced- and biased-ligand-bound states. We found that the intracellular cavity of μOR exists in an equilibrium between closed and multiple open conformations with coupled conformational changes on the transmembrane helices 3, 5, 6, and 7, and that the population of each open conformation determines the G-protein- and arrestin-mediated signaling levels in each ligand-bound state. These findings provide insight into the biased signaling of GPCRs and will be helpful for development of analgesics that stimulate μOR with reduced tolerance and dependence.
61.
Nitrogen detected TROSY at high field yields high resolution and sensitivity for protein NMR.
Show abstractDetection of (15)N in multidimensional NMR experiments of proteins has sparsely been utilized because of the low gyromagnetic ratio (γ) of nitrogen and the presumed low sensitivity of such experiments. Here we show that selecting the TROSY components of proton-attached (15)N nuclei (TROSY (15)NH) yields high quality spectra in high field magnets (>600 MHz) by taking advantage of the slow (15)N transverse relaxation and compensating for the inherently low (15)N sensitivity. The (15)N TROSY transverse relaxation rates increase modestly with molecular weight but the TROSY gain in peak heights depends strongly on the magnetic field strength. Theoretical simulations predict that the narrowest line width for the TROSY (15)NH component can be obtained at 900 MHz, but sensitivity reaches its maximum around 1.2 GHz. Based on these considerations, a (15)N-detected 2D (1)H-(15)N TROSY-HSQC ((15)N-detected TROSY-HSQC) experiment was developed and high-quality 2D spectra were recorded at 800 MHz in 2 h for 1 mM maltose-binding protein at 278 K (τc ~ 40 ns). Unlike for (1)H detected TROSY, deuteration is not mandatory to benefit (15)N detected TROSY due to reduced dipolar broadening, which facilitates studies of proteins that cannot be deuterated, especially in cases where production requires eukaryotic expression systems. The option of recording (15)N TROSY of proteins expressed in H2O media also alleviates the problem of incomplete amide proton back exchange, which often hampers the detection of amide groups in the core of large molecular weight proteins that are expressed in D2O culture media and cannot be refolded for amide back exchange. These results illustrate the potential of (15)NH-detected TROSY experiments as a means to exploit the high resolution offered by high field magnets near and above 1 GHz.
60.
Elucidation of the CCR1- and CCR5-binding modes of MIP-1α by application of an NMR spectra reconstruction method to the transferred cross-saturation experiments.
Show abstractC-C chemokine receptor 1 (CCR1) and CCR5 are involved in various inflammation and immune responses, and regulate the progression of the autoimmune diseases differently. However, the number of residues identified at the binding interface was not sufficient to clarify the differences in the CCR1- and CCR5-binding modes to MIP-1α, because the NMR measurement time for CCR1 and CCR5 samples was limited to 24 h, due to their low stability. Here we applied a recently developed NMR spectra reconstruction method, Conservation of experimental data in ANAlysis of FOuRier, to the amide-directed transferred cross-saturation experiments of chemokine receptors, CCR1 and CCR5, embedded in lipid bilayers of the reconstituted high density lipoprotein, and MIP-1α. Our experiments revealed that the residues on the N-loop and β-sheets of MIP-1α are close to both CCR1 and CCR5, and those in the C-terminal helix region are close to CCR5. These results suggest that the genetic influence of the single nucleotide polymorphisms of MIP-1α that accompany substitution of residues in the C-terminal helix region, E57 and V63, would provide clues toward elucidating how the CCR5-MIP-1α interaction affects the progress of autoimmune diseases.
59.
Structure-Based Development of a Protein-Protein Interaction Inhibitor Targeting Tumor Necrosis Factor Receptor-Associated Factor 6.
58.
Increased resolution of aromatic cross peaks using alternate 13C labeling and TROSY.
Show abstractFor typical globular proteins, contacts involving aromatic side chains would constitute the largest number of distance constraints that could be used to define the structure of proteins and protein complexes based on NOE contacts. However, the (1)H NMR signals of aromatic side chains are often heavily overlapped, which hampers extensive use of aromatic NOE cross peaks. Some of this overlap can be overcome by recording (13)C-dispersed NOESY spectra. However, the resolution in the carbon dimension is rather low due to the narrow dispersion of the carbon signals, large one-bond carbon-carbon (C-C) couplings, and line broadening due to chemical shift anisotropy (CSA). Although it has been noted that the CSA of aromatic carbons could be used in TROSY experiments for enhancing resolution, this has not been used much in practice because of complications arising from large aromatic one-bond C-C couplings, and 3D or 4D carbon dispersed NOESY are typically recorded at low resolution hampering straightforward peak assignments. Here we show that the aromatic TROSY effect can optimally be used when employing alternate (13)C labeling using 2-(13)C glycerol, 2-(13)C pyruvate, or 3-(13)C pyruvate as the carbon source. With the elimination of the strong one-bond C-C coupling, the TROSY effect can easily be exploited. We show that (1)H-(13)C TROSY spectra of alternately (13)C labeled samples can be recorded at high resolution, and we employ 3D NOESY aromatic-TROSY spectra to obtain valuable intramolecular and intermolecular cross peaks on a protein complex.
57.
Development of a method for reconstruction of crowded NMR spectra from undersampled time-domain data.
Show abstractNMR is a unique methodology for obtaining information about the conformational dynamics of proteins in heterogeneous biomolecular systems. In various NMR methods, such as transferred cross-saturation, relaxation dispersion, and paramagnetic relaxation enhancement experiments, fast determination of the signal intensity ratios in the NMR spectra with high accuracy is required for analyses of targets with low yields and stabilities. However, conventional methods for the reconstruction of spectra from undersampled time-domain data, such as linear prediction, spectroscopy with integration of frequency and time domain, and analysis of Fourier, and compressed sensing were not effective for the accurate determination of the signal intensity ratios of the crowded two-dimensional spectra of proteins. Here, we developed an NMR spectra reconstruction method, "conservation of experimental data in analysis of Fourier" (Co-ANAFOR), to reconstruct the crowded spectra from the undersampled time-domain data. The number of sampling points required for the transferred cross-saturation experiments between membrane proteins, photosystem I and cytochrome b 6 f, and their ligand, plastocyanin, with Co-ANAFOR was half of that needed for linear prediction, and the peak height reduction ratios of the spectra reconstructed from truncated time-domain data by Co-ANAFOR were more accurate than those reconstructed from non-uniformly sampled data by compressed sensing.
56.
NMR resonance assignments of the catalytic domain of human serine/threonine phosphatase calcineurin in unligated and PVIVIT-peptide-bound states.
Show abstractCalcineurin (Cn) is a serine/threonine phosphatase that plays pivotal roles in many physiological processes. In T cell, Cn targets the nuclear factors of activated T-cell (NFATs), transcription factors that activate cytokine genes. Elevated intracellular calclium concentration activates Cn to dephosphorylate multiple serine residues within the NFAT regulatory domain, which triggers joint nuclear translocation of NFAT and Cn. This relies on the interaction between the catalytic domain of Cn (CnA) and the conserved PxIxIT motif. Here, we present the assignment of CnA resonances in unligated form and in complex with a 14-residue peptide containing a PVIVIT sequence that was derived from affinity driven peptide selection based on the conserved PxIxIT motif of NFATs. Although a complete assignment was not possible mainly due to the paramagnetic line broadening induced by an iron in the CnA catalytic center, the assignment was extensively verified by amino-acid selective labeling of Arg, Leu, Lys, and Val, which cover one third of the CnA residues. Nevertheless, the assignments were used to determine the structure of the CnA-PVIVIT peptide complex and provide the basis for investigation of the interactions of CnA with physiological interaction partners and small organic compounds that disrupt the Cn-NFAT interaction.
55.
Suppression of problematic compound oligomerization by cosolubilization of nondetergent sulfobetaines.
Show abstractNumerous small organic compounds exist in equilibrium among monomers, soluble oligomers, and insoluble aggregates in aqueous solution. Compound aggregation is a major reason for false positives in drug screening, and even soluble oligomers can interfere with structural and biochemical analyses. However, an efficient way to manage the equilibrium of aggregation-prone compounds, especially those involved with soluble oligomers, has not been established. In this study, solution NMR spectroscopy was used as a suitable technique to detect compound oligomers in equilibrium, and it was demonstrated that cosolubilization of nondetergent sulfobetaines (NDSBs) can largely suppress compound oligomerization and aggregation by shifting the equilibrium toward the monomers. The rotational correlation time was obtained from the ratio of the selective and nonselective longitudinal NMR relaxation times, which directly and quantitatively reflected the apparent sizes of the compounds in the equilibrium. The rotational correlation time of the aggregation-prone compound SKF86002 (1 mM) was substantially reduced from 0.31 to 0.23 ns by cosolubilization of 100 mM NDSB195. NDSB cosolubilization allowed us to perform successful structural and biochemical experiments with substantially fewer artifacts, which represents a strategy to directly resolve the problematic oligomerization and aggregation of compounds.
2014
54.
Functional dynamics of deuterated β2 -adrenergic receptor in lipid bilayers revealed by NMR spectroscopy.
Show abstractG-protein-coupled receptors (GPCRs) exist in conformational equilibrium between active and inactive states, and the former population determines the efficacy of signaling. However, the conformational equilibrium of GPCRs in lipid bilayers is unknown owing to the low sensitivities of their NMR signals. To increase the signal intensities, a deuteration method was developed for GPCRs expressed in an insect cell/baculovirus expression system. The NMR sensitivities of the methionine methyl resonances from the β2 -adrenergic receptor (β2 AR) in lipid bilayers of reconstituted high-density lipoprotein (rHDL) increased by approximately 5-fold upon deuteration. NMR analyses revealed that the exchange rates for the conformational equilibrium of β2 AR in rHDLs were remarkably different from those measured in detergents. The timescales of GPCR signaling, calculated from the exchange rates, are faster than those of receptor tyrosine kinases and thus enable rapid neurotransmission and sensory perception.
53.
Dynamic multidrug recognition by multidrug transcriptional repressor LmrR.
Show abstract & figure
LmrR is a multidrug transcriptional repressor that controls the expression of a major multidrug transporter, LmrCD, in Lactococcus lactis. However, the molecular mechanism by which LmrR binds to structurally unrelated compounds and is released from the promoter region remains largely unknown. Here, we structurally and dynamically characterized LmrR in the apo, compound-bound and promoter-bound states. The compound-binding site of LmrR exhibits ps-μs dynamics in the apo state, and compound ligation shifts the preexisting conformational equilibrium to varying extents to achieve multidrug recognition. Meanwhile, the compound binding induces redistribution of ps-ns dynamics to the allosteric sites, which entropically favors the high-affinity recognition. Furthermore, the reciprocal compound/promoter binding by LmrR is achieved by the incompatible conformational ensembles between the compound- and promoter-bound states. Collectively, the data show how LmrR can dynamically exert its functions through promiscuous multi-target interactions, in a manner that cannot be understood by a static structural view.
52.
Allosteric enhancement of MAP kinase p38α’s activity and substrate selectivity by docking interactions.
Show abstract & figure
Mitogen-activated protein kinases (MAPKs) are essential to intracellular signal transduction. MAPKs anchor their pathway-specific substrates through so-called 'docking interactions' at locations distal from the active site. Docking interactions ensure efficient substrate recognition, but their contribution to the kinase reaction itself remains unclear. Herein, we use solution NMR to analyze the interaction between dually phosphorylated, active human p38α and the C-terminal fragments of its substrate MK2. p38α phosphorylation and ATP loading collaboratively induce the active conformation; subsequently, p38α accommodates MK2 phosphoacceptor residues in its active site. The docking interaction enhances binding of ATP and the phosphoacceptor to p38α, accelerating the phosphotransfer reaction. Thus, the docking interaction enhances p38α's enzymatic activity toward pathway-specific substrates allosterically as well as by the anchor effect. These findings clarify how MAPK cascades are organized in cells, even under ATP-depleted conditions often associated with environmental stress.
51.
The LxVP and PxIxIT NFAT motifs bind jointly to overlapping epitopes on calcineurin’s catalytic domain distant to the regulatory domain.
Show abstractThe serine/threonine phosphatase calcineurin (Cn) targets the nuclear factors of activated T cells (NFATs) that activate cytokine genes. Calcium influx activates Cn to dephosphorylate multiple serine residues within the ∼200 residue NFAT regulatory domain, which triggers joint nuclear translocation of NFAT and Cn. The dephosphorylation process relies on the interaction between Cn and the conserved motifs PxIxIT and LxVP, which are located N- and C-terminal to the phosphorylation sites in NFAT's regulatory domain. Here, we show that an NFATc1-derived 15-residue peptide segment containing the conserved LxVP motif binds to an epitope on Cn's catalytic domain (CnA), which overlaps with the previously established PxIxIT binding site on CnA and is distant to the regulatory domain (CnB). Both NFAT motifs partially compete for binding but do not fully displace each other on the CnA epitope, revealing that both segments bind simultaneously to the same epitope on the catalytic domain.
50.
Cross-saturation and transferred cross-saturation experiments.
49.
Functional dynamics of cell surface membrane proteins.
48.
Large Protein Complexes Revealed by Solution-State NMR: G Proteins and G Protein-Activated Inwardly Rectifying Potassium Ion Channel
Advances in Biological Solid-State NMR: Proteins and Membrane-Active Peptides 2014; Chapter 26:501-532
47.
Structure-based approach to improve a small-molecule inhibitor by the use of a competitive peptide ligand.
Show abstractStructural information about the target-compound complex is invaluable in the early stage of drug discovery. In particular, it is important to know into which part of the initial compound additional interaction sites could be introduced to improve its affinity. Herein, we demonstrate that the affinity of a small-molecule inhibitor for its target protein could be successfully improved by the constructive introduction of the interaction mode of a competitive peptide. The strategy involved the discrimination of overlapping and non-overlapping peptide-compound pharmacophores by the use of a ligand-based NMR spectroscopic approach, INPHARMA. The obtained results enabled the design of a new compound with improved affinity for the platelet receptor glycoprotein VI (GPVI). The approach proposed herein efficiently combines the advantages of compounds and peptides for the development of higher-affinity druglike ligands.
46.
Degradation of activated K-Ras orthologue via K-Ras-specific lysine residues is required for cytokinesis.
Show abstractMammalian cells encode three closely related Ras proteins, H-Ras, N-Ras, and K-Ras. Oncogenic K-Ras mutations frequently occur in human cancers, which lead to dysregulated cell proliferation and genomic instability. However, mechanistic role of the Ras isoform regulation have remained largely unknown. Furthermore, the dynamics and function of negative regulation of GTP-loaded K-Ras have not been fully investigated. Here, we demonstrate RasG, the Dictyostelium orthologue of K-Ras, is targeted for degradation by polyubiquitination. Both ubiquitination and degradation of RasG were strictly associated with RasG activity. High resolution tandem mass spectrometry (LC-MS/MS) analysis indicated that RasG ubiquitination occurs at C-terminal lysines equivalent to lysines found in human K-Ras but not in H-Ras and N-Ras homologues. Substitution of these lysine residues with arginines (4KR-RasG) diminished RasG ubiquitination and increased RasG protein stability. Cells expressing 4KR-RasG failed to undergo proper cytokinesis and resulted in multinucleated cells. Ectopically expressed human K-Ras undergoes polyubiquitin-mediated degradation in Dictyostelium, whereas human H-Ras and a Dictyostelium H-Ras homologue (RasC) are refractory to ubiquitination. Our results indicate the existence of GTP-loaded K-Ras orthologue-specific degradation system in Dictyostelium, and further identification of the responsible E3-ligase may provide a novel therapeutic approach against K-Ras-mutated cancers.
45.
Identification of a binding element for the cytoplasmic regulator FROUNT in the membrane-proximal C-terminal region of chemokine receptors CCR2 and CCR5.
2013
44.
Rapid identification of ligand-binding sites by using an assignment-free NMR approach.
43.
Perdeuteration and methyl-selective (1)H, (13)C-labeling by using a Kluyveromyces lactis expression system.
Show abstractThe production of stable isotope-labeled proteins is critical in structural analyses of large molecular weight proteins using NMR. Although prokaryotic expression systems using Escherichia coli have been widely used for this purpose, yeast strains have also been useful for the expression of functional eukaryotic proteins. Recently, we reported a cost-effective stable isotope-labeled protein expression using the hemiascomycete yeast Kluyveromyces lactis (K. lactis), which allow us to express exogenous proteins at costs comparable to prokaryotic expression systems. Here, we report the successful production of highly deuterated (>90 %) protein in the K. lactis system. We also examined the methyl-selective (1)H, (13)C-labeling of Ile, Leu, and Val residues using commonly used amino acid precursors. The efficiency of (1)H-(13)C-incorporation varied significantly based on the amino acid. Although a high level of (1)H-(13)C-incorporation was observed for the Ile δ1 position, (1)H, (13)C-labeling rates of Val and Leu methyl groups were limited due to the mitochondrial localization of enzymes involved in amino acid biosynthesis and the lack of transporters for α-ketoisovalerate in the mitochondrial membrane. In line with this notion, the co-expression with branched-chain-amino-acid aminotransferase in the cytosol significantly improved the incorporation rates of amino acid precursors. Although it would be less cost-effective, addition of (13)C-labeled valine can circumvent problems associated with precursors and achieve high level (1)H, (13)C-labeling of Val and Leu. Taken together, the K. lactis system would be a good alternative for expressing large eukaryotic proteins that need deuteration and/or the methyl-selective (1)H, (13)C-labeling for the sensitive detection of NMR resonances.
42.
A homogeneous, high-throughput assay for phosphatidylinositol 5-phosphate 4-kinase with a novel, rapid substrate preparation.
Show abstractPhosphoinositide kinases regulate diverse cellular functions and are important targets for therapeutic development for diseases, such as diabetes and cancer. Preparation of the lipid substrate is crucial for the development of a robust and miniaturizable lipid kinase assay. Enzymatic assays for phosphoinositide kinases often use lipid substrates prepared from lyophilized lipid preparations by sonication, which result in variability in the liposome size from preparation to preparation. Herein, we report a homogeneous 1536-well luciferase-coupled bioluminescence assay for PI5P4Kα. The substrate preparation is novel and allows the rapid production of a DMSO-containing substrate solution without the need for lengthy liposome preparation protocols, thus enabling the scale-up of this traditionally difficult type of assay. The Z'-factor value was greater than 0.7 for the PI5P4Kα assay, indicating its suitability for high-throughput screening applications. Tyrphostin AG-82 had been identified as an inhibitor of PI5P4Kα by assessing the degree of phospho transfer of γ-(32)P-ATP to PI5P; its inhibitory activity against PI5P4Kα was confirmed in the present miniaturized assay. From a pilot screen of a library of bioactive compounds, another tyrphostin, I-OMe tyrphostin AG-538 (I-OMe-AG-538), was identified as an ATP-competitive inhibitor of PI5P4Kα with an IC(50) of 1 µM, affirming the suitability of the assay for inhibitor discovery campaigns. This homogeneous assay may apply to other lipid kinases and should help in the identification of leads for this class of enzymes by enabling high-throughput screening efforts.
2012
41.
Functional equilibrium of the KcsA structure revealed by NMR.
Show abstractKcsA is a tetrameric K(+) channel that is activated by acidic pH. Under open conditions of the helix bundle crossing, the selectivity filter undergoes an equilibrium between permeable and impermeable conformations. Here we report that the population of the permeable conformation (p(perm)) positively correlates with the tetrameric stability and that the population in reconstituted high density lipoprotein, where KcsA is surrounded by the lipid bilayer, is lower than that in detergent micelles, indicating that dynamic properties of KcsA are different in these two media. Perturbation of the membrane environment by the addition of 1-3% 2,2,2-trifluoroethanol increases p(perm) and the open probability, revealed by NMR and single-channel recording analyses. These results demonstrate that KcsA inactivation is determined not only by the protein itself but also by the surrounding membrane environments.
40.
Functional dynamics of proteins revealed by solution NMR.
Show abstractSolution NMR spectroscopy can analyze the dynamics of proteins on a wide range of timescales, from picoseconds to even days, in a site-specific manner, and thus its results are complementary to the detailed but largely static structural information obtained by X-ray crystallography. We review recent progresses in a variety of NMR techniques, including relaxation dispersion and paramagnetic relaxation enhancement (PRE), that permit the observation of the low-populated states, which had been 'invisible' with other techniques. In addition, we review how NMR spectroscopy can be used to elucidate functionally relevant protein dynamics.
39.
Structural basis for the Golgi association by the pleckstrin homology domain of the ceramide trafficking protein (CERT).
Show abstractCeramide transport from the endoplasmic reticulum to the Golgi apparatus is crucial in sphingolipid biosynthesis, and the process relies on the ceramide trafficking protein (CERT), which contains pleckstrin homology (PH) and StAR-related lipid transfer domains. The CERT PH domain specifically recognizes phosphatidylinositol 4-monophosphate (PtdIns(4)P), a characteristic phosphoinositide in the Golgi membrane, and is indispensable for the endoplasmic reticulum-to-Golgi transport of ceramide by CERT. In this study, we determined the three-dimensional structure of the CERT PH domain by using solution NMR techniques. The structure revealed the presence of a characteristic basic groove near the canonical PtdIns(4)P recognition site. An extensive interaction study using NMR and other biophysical techniques revealed that the basic groove coordinates the CERT PH domain for efficient PtdIns(4)P recognition and localization in the Golgi apparatus. The notion was also supported by Golgi mislocalization of the CERT mutants in living cells. The distinctive binding modes reflect the functions of PH domains, as the basic groove is conserved only in the PH domains involved with the PtdIns(4)P-dependent lipid transport activity but not in those with the signal transduction activity.
38.
Low-γ Nuclei Detection Experiments for Biomolecular NMR
Recent Developments in Biomolecular NMR 2012; Chapter 2:25-52
37.
Efficacy of the β₂-adrenergic receptor is determined by conformational equilibrium in the transmembrane region.
Nat Commun 2012; 3:1045
Show abstractMany drugs that target G-protein-coupled receptors (GPCRs) induce or inhibit their signal transduction with different strengths, which affect their therapeutic properties. However, the mechanism underlying the differences in the signalling levels is still not clear, although several structures of GPCRs complexed with ligands determined by X-ray crystallography are available. Here we utilized NMR to monitor the signals from the methionine residue at position 82 in neutral antagonist- and partial agonist-bound states of β(2)-adrenergic receptor (β(2)AR), which are correlated with the conformational changes of the transmembrane regions upon activation. We show that this residue exists in a conformational equilibrium between the inverse agonist-bound states and the full agonist-bound state, and the population of the latter reflects the signal transduction level in each ligand-bound state. These findings provide insights into the multi-level signalling of β(2)AR and other GPCRs, including the basal activity, and the mechanism of signal transduction mediated by GPCRs.
2011
36.
Speeding up direct (15)N detection: hCaN 2D NMR experiment.
Show abstractExperiments detecting low gyromagnetic nuclei have recently been proposed to utilize the relatively slow relaxation properties of these nuclei in comparison to (1)H. Here we present a new type of (15)N direct-detection experiment. Like the previously proposed CaN experiment (Takeuchi et al. in J Biomol NMR 47:271-282, 2010), the hCaN experiment described here sequentially connects amide (15)N resonances, but utilizes the initial high polarization and the faster recovery of the (1)H nucleus to shorten the recycling delay. This allows recording 2D (15)N-detected NMR experiments on proteins within a few hours, while still obtaining superior resolution for (13)C and (15)N, establishing sequential assignments through prolines, and at conditions where amide protons exchange rapidly. The experiments are demonstrated on various biomolecules, including the small globular protein GB1, the 22 kDa HEAT2 domain of eIF4G, and an unstructured polypeptide fragment of NFAT1, which contains many SerPro sequence repeats.
35.
Protein-ligand docking guided by ligand pharmacophore-mapping experiment by NMR.
Show abstractWe developed a new protein-ligand docking calculation method using experimental NMR data. Recently, we proposed a novel ligand epitope-mapping experiment, which utilizes the difference between the longitudinal relaxation rates of ligand protons with and without irradiation of target protein protons (DIRECTION epitope-mapping experiment; Y. Mizukoshi, et al., An accurate pharmacophore mapping method by NMR, submitted for publication). Although the epitope-mapping experiment is simple and rapid, the result should reflect the proximity of ligand protons to the target protein surface. However, it cannot directly provide the protein-ligand complex structure without any other structural information. While the accuracy of protein-ligand docking software is insufficient, the software can provide many candidate complex structures. In many cases, the correct complex structure is included in the set of predicted complex structures and the correct structures could be selected by applying the above experimental result of ligand epitope mapping. In the current study, we combined the protein-ligand docking software with the NMR experimental information so as to improve the prediction of the protein-ligand complex structure. Consequently, the prediction accuracy was improved by 1.3-1.9 times (from ca. 50% to ca. 70%) in a self-docking test for the simulated epitope mapping result. Moreover, this method was applied to actual NMR experiments, and it successfully reconstructed the protein-ligand complex structures.
34.
Structure of the VP16 transactivator target in the Mediator.
Show abstractThe human Mediator coactivator complex interacts with many transcriptional activators and facilitates recruitment of RNA polymerase II to promote target gene transcription. The MED25 subunit is a critical target of the potent herpes simplex 1 viral transcriptional activator VP16. Here we determine the solution structure of the MED25 VP16-binding domain (VBD) and define its binding site for the N-terminal portion of the VP16 transactivation domain (TADn). A hydrophobic furrow, formed by a β-barrel and two α-helices in MED25 VBD, interacts tightly with VP16 TADn. Mutations in this furrow prevent binding of VP16 TAD to MED25 VBD and interfere with the ability of overexpressed MED25 VBD to inhibit VP16-dependent transcriptional activation in vivo. This detailed molecular understanding of transactivation by the benchmark activator VP16 could provide important insights into viral and cellular gene activation mechanisms.
33.
Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors.
Show abstractThe guanosine triphosphate (GTP)--loaded form of the guanosine triphosphatase (GTPase) Ras initiates multiple signaling pathways by binding to various effectors, such as the kinase Raf and phosphatidylinositol 3-kinase (PI3K). Ras activity is increased by guanine nucleotide exchange factors that stimulate guanosine diphosphate release and GTP loading and is inhibited by GTPase-activating proteins that stimulate GTP hydrolysis. KRAS is the most frequently mutated RAS gene in cancer. Here, we report that monoubiquitination of lysine-147 in the guanine nucleotide-binding motif of wild-type K-Ras could lead to enhanced GTP loading. Furthermore, ubiquitination increased the binding of the oncogenic Gly12Val mutant of K-Ras to the downstream effectors PI3K and Raf. Thus, monoubiquitination could enhance GTP loading on K-Ras and increase its affinity for specific downstream effectors, providing a previously unidentified mechanism for Ras activation.
32.
NMR analyses of the Gbetagamma binding and conformational rearrangements of the cytoplasmic pore of G protein-activated inwardly rectifying potassium channel 1 (GIRK1).
Show abstractG protein-activated inwardly rectifying potassium channel (GIRK) plays crucial roles in regulating heart rate and neuronal excitability in eukaryotic cells. GIRK is activated by the direct binding of heterotrimeric G protein βγ subunits (Gβγ) upon stimulation of G protein-coupled receptors, such as M2 acetylcholine receptor. The binding of Gβγ to the cytoplasmic pore (CP) region of GIRK causes structural rearrangements, which are assumed to open the transmembrane ion gate. However, the crucial residues involved in the Gβγ binding and the structural mechanism of GIRK gating have not been fully elucidated. Here, we have characterized the interaction between the CP region of GIRK and Gβγ, by ITC and NMR. The ITC analyses indicated that four Gβγ molecules bind to a tetramer of the CP region of GIRK with a dissociation constant of 250 μM. The NMR analyses revealed that the Gβγ binding site spans two neighboring subunits of the GIRK tetramer, which causes conformational rearrangements between subunits. A possible binding mode and mechanism of GIRK gating are proposed.
31.
HNCA-TOCSY-CANH experiments with alternate (13)C- (12)C labeling: a set of 3D experiment with unique supra-sequential information for mainchain resonance assignment.
Show abstractDescribed here is a set of three-dimensional (3D) NMR experiments that rely on CACA-TOCSY magnetization transfer via the weak ³J(CαCα) coupling. These pulse sequences, which resemble recently described (13)C detected CACA-TOCSY (Takeuchi et al. 2010) experiments, are recorded in (1)H(2)O, and use (1)H excitation and detection. These experiments require alternate (13)C-(12)C labeling together with perdeuteration, which allows utilizing the small ³J(CαCα) scalar coupling that is otherwise masked by the stronger (1)J(CC) couplings in uniformly (13)C labeled samples. These new experiments provide a unique assignment ladder-mark that yields bidirectional supra-sequential information and can readily straddle proline residues. Unlike the conventional HNCA experiment, which contains only sequential information to the ¹³C(α) of the preceding residue, the 3D hnCA-TOCSY-caNH experiment can yield sequential correlations to alpha carbons in positions i-1, i + 1 and i-2. Furthermore, the 3D hNca-TOCSY-caNH and Hnca-TOCSY-caNH experiments, which share the same magnetization pathway but use a different chemical shift encoding, directly couple the (15)N-(1)H spin pair of residue i to adjacent amide protons and nitrogens at positions i-2, i-1, i + 1 and i + 2, respectively. These new experimental features make protein backbone assignments more robust by reducing the degeneracy problem associated with the conventional 3D NMR experiments.
2010
30.
Distinctive CD3 heterodimeric ectodomain topologies maximize antigen-triggered activation of alpha beta T cell receptors.
29.
Nitrogen-detected CAN and CON experiments as alternative experiments for main chain NMR resonance assignments.
Show abstractHeteronuclear direct-detection experiments, which utilize the slower relaxation properties of low gamma nuclei, such as (13)C have recently been proposed for sequence-specific assignment and structural analyses of large, unstructured, and/or paramagnetic proteins. Here we present two novel (15)N direct-detection experiments. The CAN experiment sequentially connects amide (15)N resonances using (13)C(alpha) chemical shift matching, and the CON experiment connects the preceding (13)C' nuclei. When starting from the same carbon polarization, the intensities of nitrogen signals detected in the CAN or CON experiments would be expected four times lower than those of carbon resonances observed in the corresponding (13)C-detecting experiment, NCA-DIPAP or NCO-IPAP (Bermel et al. 2006b; Takeuchi et al. 2008). However, the disadvantage due to the lower gamma is counteracted by the slower (15)N transverse relaxation during detection, the possibility for more efficient decoupling in both dimensions, and relaxation optimized properties of the pulse sequences. As a result, the median S/N in the (15)N observe CAN experiment is 16% higher than in the (13)C observe NCA-DIPAP experiment. In addition, significantly higher sensitivity was observed for those residues that are hard to detect in the NCA-DIPAP experiment, such as Gly, Ser and residues with high-field C(alpha) resonances. Both CAN and CON experiments are able to detect Pro resonances that would not be observed in conventional proton-detected experiments. In addition, those experiments are free from problems of incomplete deuterium-to-proton back exchange in amide positions of perdeuterated proteins expressed in D(2)O. Thus, these features and the superior resolution of (15)N-detected experiments provide an attractive alternative for main chain assignments. The experiments are demonstrated with the small model protein GB1 at conditions simulating a 150 kDa protein, and the 52 kDa glutathione S-transferase dimer, GST.
28.
Autoinhibitory interaction in the multidomain adaptor protein Nck: possible roles in improving specificity and functional diversity.
Show abstractNck is a functionally versatile multidomain adaptor protein consisting of one SH2 and three SH3 domains. In most cases, the SH2 domain mediates binding to tyrosine-phosphorylated receptors or cytosolic proteins, which leads to the formation of larger protein complexes via the SH3 domains. Nck plays a pivotal role in T-cell receptor-mediated reorganization of the actin cytoskeleton as well as in the formation of the immunological synapses. The modular domain structure and the functionality of the individual domains suggest that they might act independently. Here we report an interesting intramolecular interaction within Nck that occurs between a noncanonical yet conserved (K/R)x(K/R)RxxS sequence in the linker between the first and second SH3 domain (SH3.1/SH3.2) and the second SH3 domain (SH3.2). Because this interaction masks the proline-rich sequence binding site of the SH3.2 domain, the intramolecular interaction is self-inhibitory. This intramolecular interaction could, at least partially, explain the remarkable specificity of Nck toward proteins with proline-rich sequences. It may prevent nonspecific low-affinity binding while keeping the site available for high-affinity bivalent ligands that can bind multiple sites in Nck. This indicates that Nck does not simply adopt a "beads on a string" architecture but incorporates a higher-order organization for improved specificity and functionality.
27.
NMR analyses of the interaction between CCR5 and its ligand using functional reconstitution of CCR5 in lipid bilayers.
26.
CACA-TOCSY with alternate 13C-12C labeling: a 13Calpha direct detection experiment for mainchain resonance assignment, dihedral angle information, and amino acid type identification.
Show abstract & figure
We present a (13)C direct detection CACA-TOCSY experiment for samples with alternate (13)C-(12)C labeling. It provides inter-residue correlations between (13)C(alpha) resonances of residue i and adjacent C(alpha)s at positions i - 1 and i + 1. Furthermore, longer mixing times yield correlations to C(alpha) nuclei separated by more than one residue. The experiment also provides C(alpha)-to-sidechain correlations, some amino acid type identifications and estimates for psi dihedral angles. The power of the experiment derives from the alternate (13)C-(12)C labeling with [1,3-(13)C] glycerol or [2-(13)C] glycerol, which allows utilizing the small scalar (3)J(CC) couplings that are masked by strong (1)J(CC) couplings in uniformly (13)C labeled samples.
25.
Structural basis underlying the dual gate properties of KcsA.
Show abstractKcsA is a prokaryotic pH-dependent potassium (K) channel. Its activation, by a decrease in the intracellular pH, is coupled with its subsequent inactivation, but the underlying mechanisms remain elusive. Here, we have investigated the conformational changes and equilibrium of KcsA by using solution NMR spectroscopy. Controlling the temperature and pH of KcsA samples produced three distinct methyl-TROSY and NOESY spectra, corresponding to the resting, activated, and inactivated states. The pH-dependence of the signals from the extracellular side was affected by the mutation of H25 on the intracellular side, indicating the coupled conformational changes of the extracellular and intracellular gates. K(+) titration and NOE experiments revealed that the inactivated state was obtained by the replacement of K(+) with H(2)O, which may interfere with the K(+)-permeation. This structural basis of the activation-coupled inactivation is closely related to the C-type inactivation of other K channels.
24.
High-resolution 3D CANCA NMR experiments for complete mainchain assignments using C(alpha) direct detection.
23.
Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data.
2009
22.
Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4.
Show abstractThe chemokine stromal cell-derived factor-1 (SDF-1/CXCL12) and its G-protein-coupled receptor (GPCR) CXCR4 play fundamental roles in many physiological processes, and CXCR4 is a drug target for various diseases such as cancer metastasis and human immunodeficiency virus, type 1, infection. However, almost no structural information about the SDF-1-CXCR4 interaction is available, mainly because of the difficulties in expression, purification, and crystallization of CXCR4. In this study, an extensive investigation of the preparation of CXCR4 and optimization of the experimental conditions enables NMR analyses of the interaction between the full-length CXCR4 and SDF-1. We demonstrated that the binding of an extended surface on the SDF-1 beta-sheet, 50-s loop, and N-loop to the CXCR4 extracellular region and that of the SDF-1 N terminus to the CXCR4 transmembrane region, which is critical for G-protein signaling, take place independently by methyl-utilizing transferred cross-saturation experiments along with the usage of the CXCR4-selective antagonist AMD3100. Furthermore, based upon the data, we conclude that the highly dynamic SDF-1 N terminus in the 1st step bound state plays a crucial role in efficiently searching the deeply buried binding pocket in the CXCR4 transmembrane region by the "fly-casting" mechanism. This is the first structural analyses of the interaction between a full-length GPCR and its chemokine, and our methodology would be applicable to other GPCR-ligand systems, for which the structural studies are still challenging.
21.
The alphabeta T cell receptor is an anisotropic mechanosensor.
Show abstractThymus-derived lymphocytes protect mammalian hosts against virus- or cancer-related cellular alterations through immune surveillance, eliminating diseased cells. In this process, T cell receptors (TCRs) mediate both recognition and T cell activation via their dimeric alphabeta, CD3 epsilon gamma, CD3 epsilon delta, and CD3 zeta zeta subunits using an unknown structural mechanism. Here, site-specific binding topology of anti-CD3 monoclonal antibodies (mAbs) and dynamic TCR quaternary change provide key clues. Agonist mAbs footprint to the membrane distal CD3 epsilon lobe that they approach diagonally, adjacent to the lever-like C beta FG loop that facilitates antigen (pMHC)-triggered activation. In contrast, a non-agonist mAb binds to the cleft between CD3 epsilon and CD3 gamma in a perpendicular mode and is stimulatory only subsequent to an external tangential but not a normal force ( approximately 50 piconewtons) applied via optical tweezers. Specific pMHC but not irrelevant pMHC activates a T cell upon application of a similar force. These findings suggest that the TCR is an anisotropic mechanosensor, converting mechanical energy into a biochemical signal upon specific pMHC ligation during immune surveillance. Activating anti-CD3 mAbs mimic this force via their intrinsic binding mode. A common TCR quaternary change rather than conformational alterations can better facilitate structural signal initiation, given the vast array of TCRs and their specific pMHC ligands.
20.
An NMR method for the determination of protein binding interfaces using TEMPOL-induced chemical shift perturbations.
Show abstractThe determination of protein-protein interfaces is of crucial importance to understand protein function and to guide the design of compounds. To identify protein-protein interface by NMR spectroscopy, 13C NMR paramagnetic shifts induced by freely diffusing 4-hydroxy-2, 2, 6, 6-tetramethyl-piperidine-1-oxyl (TEMPOL) are promising, because TEMPOL affects distinct 13C NMR chemical shifts of the solvent accessible nuclei belonging to proteins of interest, while 13C nuclei within the interior of the proteins may be distinguished by a lack of such shifts. 19.
Backbone resonance assignments for the cytoplasmic regions of G protein-activated inwardly rectifying potassium channel 1 (GIRK1).
Show abstractG protein-activated inwardly rectifying potassium channel (GIRK) plays crucial roles in regulating heart rate and neuronal excitability in eukaryotic cells. A variety of ligands, including heterotrimeric G protein betagamma subunits (G betagamma), bind to the cytoplasmic regions of GIRK and modulate its activity. We established the backbone resonance assignments of (2)H/(13)C/(15)N-labeled cytoplasmic regions of mouse GIRK1, which form a tetramer with a molecular weight of 96 K.
18.
High-throughput screening of optimal solution conditions for structural biological studies by fluorescence correlation spectroscopy.
2008
17.
Alternate 13C-12C labeling for complete mainchain resonance assignments using C alpha direct-detection with applicability toward fast relaxing protein systems.
16.
Amino acid selective cross-saturation method for identification of proximal residue pairs in a protein-protein complex.
15.
Cross-saturation and transferred cross-saturation experiments
Prog Nucl Magn Reson Spectrosc 2008; 54(2):123-140
14.
Structural and functional evidence that Nck interaction with CD3epsilon regulates T-cell receptor activity.
2007
13.
Identification and characterization of the slowly exchanging pH-dependent conformational rearrangement in KcsA.
Show abstract & figure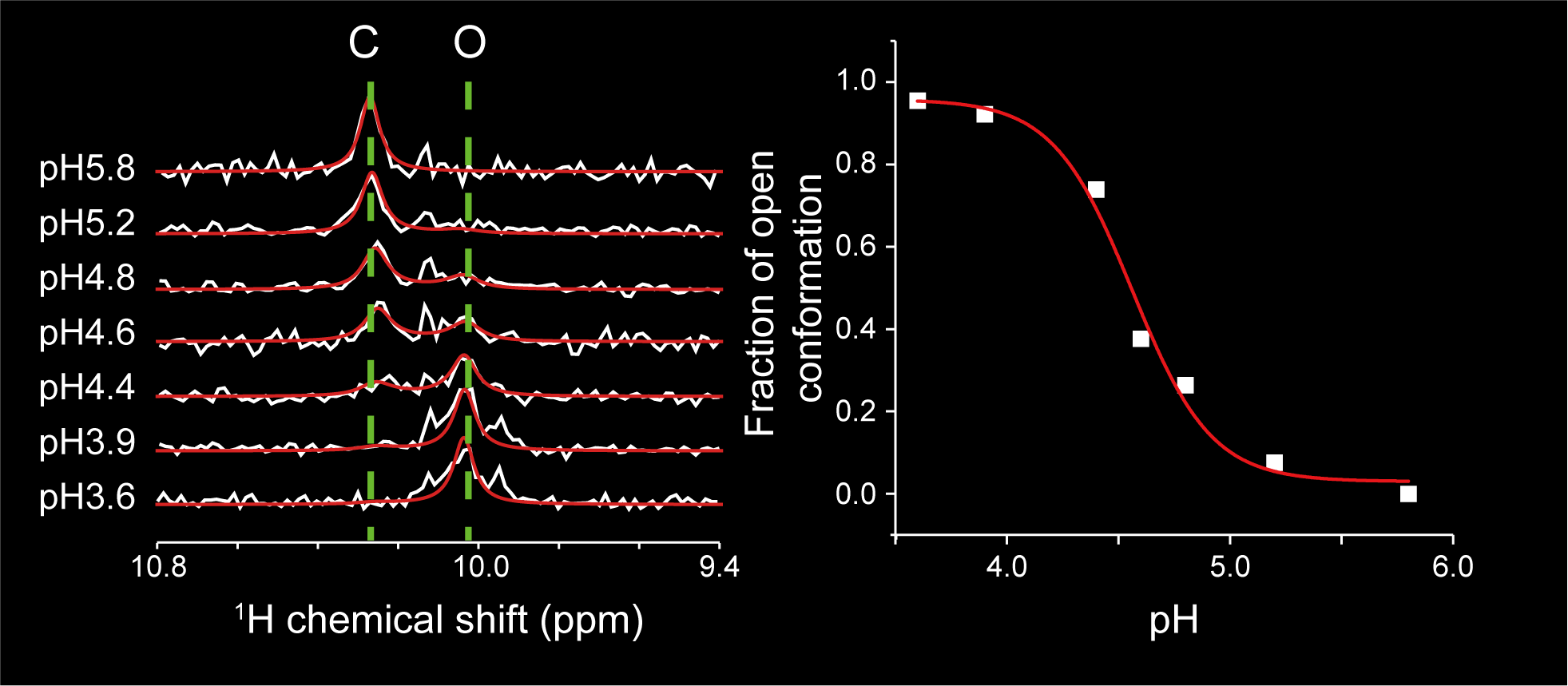
Gating of ion channels is strictly regulated by physiological conditions as well as intra/extracellular ligands. To understand the underlying structures mediating ion channel gating, we investigated the pH-dependent gating of the K(+) channel KcsA under near-physiological conditions, using solution-state NMR. In a series of (1)H(15)N-TROSY HSQC (transverse relaxation optimized spectroscopy-heteronuclear single quantum coherence) spectra measured at various pH values, significant chemical shift changes were detected between pH 3.9 and 5.2, reflecting a conformational rearrangement associated with the gating. The pH-dependent chemical shift changes were mainly observed for the resonances from the residues near the intracellular helix bundle, which has been considered to form the primary gate in the K(+) channel, as well as the intracellular extension of the inner helix. The substitution of His-25 by Ala abolished this pH-dependent conformational rearrangement, indicating that the residue serves as a "pH-sensor" for the channel. Although the electrophysiological open probability of KcsA is less than 10%, the conformations of the intracellular helix bundle between the acidic and neutral conditions seem to be remarkably different. This supports the recently proposed "dual gating" properties of the K(+) channel, in which the activation-coupled inactivation at the selectivity filter determines the channel open probability of the channel. Indeed, a pH-dependent chemical shift change was also observed for the signal from the Trp-67 indole, which is involved in a hydrogen bond network related to the activation-coupled inactivation. The slow kinetic parameter obtained for the intracellular bundle seems to fit better into the time scale for burst duration than very fast fluctuations within a burst period, indicating the existence of another gating element with faster kinetic properties.
12.
Structure of the calcineurin-NFAT complex: defining a T cell activation switch using solution NMR and crystal coordinates.
11.
1-13C amino acid selective labeling in a 2H15N background for NMR studies of large proteins.
2006
10.
Solution structure of the first SRC homology 3 domain of human Nck2.
9.
NMR studies of protein interactions.
Show abstractInteractions of proteins with other macromolecules or small molecules play important roles in most biological processes. Often, such interactions are weak and transient, and the complexes do not easily crystallize. NMR spectroscopy has the unique ability to retrieve information about these interactions and is increasingly used. Recent methodological developments have helped characterize weak protein interactions, and have in particular been applied to the study of proteins that are mostly unfolded alone but form well-defined complexes upon interaction. In addition, NMR methods have been applied to the identification and characterization of small chemicals that inhibit protein function, a primary objective of rational drug design.
2005
8.
Backbone resonance assignments for the Fv fragment of catalytic antibody 6D9 complexed with a transition state analogue
Show abstractThe catalytic antibody 6D9, which was induced by immunization with chloramphenicol phosphonate (a transition-state analogue, TSA, for the reaction), catalyzes the hydrolysis of a non-bioactive chloramphenicol monoester to generate the chloramphenicol (Miyashita et al., 1993). To understand the structurecatalytic activity relations of 6D9, we carried out NMR analyses of an Fv fragment of 6D9, which consists of a 114-residue light chain (VL) and a 122-residue heavy chain (VH), complexed with TSA. For the assignments 2D and 3D heteronuclear NMR experiments with VL- and VH-selectively 2H/13C/15N labeledFvs were used. The extent of backbone resonance assignments is 99% and 84% for VL and VH, respectively. BMRB deposit with accession number 6785.
7.
Direct determination of a membrane-peptide interface using the nuclear magnetic resonance cross-saturation method.
6.
Bead-linked proteoliposomes: a reconstitution method for nmr analyses of membrane protein-ligand interactions.
5.
[Structural basis of the K+ channel inhibition by pore-blocking toxins, revealed by NMR].
Tanpakushitsu Kakusan Koso 2005; 50(10 Suppl):1297-302
2004
4.
Channel-forming membrane permeabilization by an antibacterial protein, sapecin: determination of membrane-buried and oligomerization surfaces by NMR.
Show abstractThe action mechanism of sapecin, an antibacterial peptide with membrane permeabilization activity, was investigated. The dose dependence of the membrane permeabilization caused by sapecin was sigmoidal, suggesting that sapecin oligomerization leads to the membrane permeabilization. Solution nuclear magnetic resonance analysis of the sapecin-phospholipid vesicle complex revealed the surface buried in the membrane and oligomerization surface on the sapecin molecule. The membrane-buried surface of sapecin was determined by observing the transferred cross-saturation phenomena from the alkyl chains of the phospholipid vesicle to the amide protons of sapecin. The membrane-buried surface contains basic and highly exposed hydrophobic residues, which are suitable for interacting with the acidic bacterial membrane. The oligomerization surface was also identified by comparisons between the results from hydrogen-deuterium exchange experiments and transferred cross-saturation experiments. On the basis of the results from the NMR experiments we built a putative model of sapecin oligomers, which provides insights into the membrane permeabilization caused by insect defensins.
3.
Molecular basis of the high-affinity activation of type 1 ryanodine receptors by imperatoxin A.
Show abstractBoth imperatoxin A (IpTx(a)), a 33-residue peptide toxin from scorpion venom, and peptide A, derived from the II-III loop of dihydropyridine receptor (DHPR), interact specifically with the skeletal ryanodine receptor (RyR1), which is a Ca(2+)-release channel in the sarcoplasmic reticulum, but with considerably different affinities. IpTx(a) activates RyR1 with nanomolar affinity, whereas peptide A activates RyR1 at micromolar concentrations. To investigate the molecular basis for high-affinity activation of RyR1 by IpTx(a), we have determined the NMR solution structure of IpTx(a), and identified its functional surface by using alanine-scanning analogues. A detailed comparison of the functional surface profiles for two peptide activators revealed that IpTx(a) exhibits a large functional surface area (approx. 1900 A(2), where 1 A=0.1 nm), based on a short double-stranded antiparallel beta-sheet structure, while peptide A bears a much smaller functional surface area (approx. 800 A(2)), with the five consecutive basic residues (Arg(681), Lys(682), Arg(683), Arg(684) and Lys(685)) being clustered at the C-terminal end of the alpha-helix. The functional surface of IpTx(a) is composed of six essential residues (Leu(7), Lys(22), Arg(23), Arg(24), Arg(31) and Arg(33)) and several other important residues (His(6), Lys(8), Arg(9), Lys(11), Lys(19), Lys(20), Gly(25), Thr(26), Asn(27) and Lys(30)), indicating that amino acid residues involved in RyR1 activation make up over the half of the toxin molecule with the exception of cysteine residues. Taken together, these results suggest that the site where peptide A binds to RyR1 belongs to a subset of macrosites capable of being occupied by IpTx(a), resulting in differing the affinity and the mode of activation.
2003
2.
Structural basis of the KcsA K(+) channel and agitoxin2 pore-blocking toxin interaction by using the transferred cross-saturation method.
Show abstractWe have determined the binding site on agitoxin2 (AgTx2) to the KcsA K(+) channel by a transferred cross-saturation (TCS) experiment. The residues significantly affected in the TCS experiments formed a contiguous surface on AgTx2, and substitutions of the surface residues decreased the binding affinity to the KcsA K(+) channel. Based on properties of the AgTx2 binding site with the KcsA K(+) channel, we present a surface motif that is observed in pore-blocking toxins affecting the K(+) channel. Furthermore, we also explain the structural basis of the specificity of the K(+) channel to the toxins. The TCS method utilized here is applicable not only for the channels, which are complexed with other inhibitors, but also with a variety of regulatory molecules, and provides important information about their interface in solution.
2002
1.
Solution structure of omega-grammotoxin SIA, a gating modifier of P/Q and N-type Ca(2+) channel.
Show abstractomega-Grammotoxin SIA (GrTx) is a 36 amino acid residue protein toxin from spider venom that inhibits P/Q and N-type voltage-gated Ca(2+) channels by modifying voltage-dependent gating. We determined the three-dimensional structure of GrTx using NMR spectroscopy. The toxin adopts an "inhibitor cystine knot" motif composed of two beta-strands (Leu19-Cys21 and Cys30-Trp32) and a beta-bulge (Trp6, Gly7-Cys30) with a +2x, -1 topology, which are connected by four chain reversals. Although GrTx was originally identified as an inhibitor of voltage-gated Ca(2+) channel, it also binds to K(+) channels with lower affinity. A similar cross-reaction was observed for Hanatoxin1 (HaTx), which binds to the voltage-sensing domains of K(+) and Ca(2+) channels with different affinities. A detailed comparison of the GrTx and HaTx structures identifies a conserved face containing a large hydrophobic patch surrounded by positively charged residues. The slight differences in the surface shape, which result from the orientation of the surface aromatic residues and/or the distribution of the charged residues, may explain the differences in the binding affinity of these gating modifiers with different voltage-gated ion channels.